1.本发明涉及一种分离柴油中含硫化合物及含氮化合物的方法,具体地说,涉及一种快速地为后续测定柴油含硫及含氮化合物提供实验样品而利用固相萃取方法将柴油样品分离为饱和烃、芳烃、含硫化合物和含氮化合物的预处理方法。
背景技术:
2.柴油馏分中含硫及含氮化合物的存在会对柴油的加工、使用和存储带来一系列的危害。例如,在加工过程中柴油馏分中的含氮化合物会造成催化剂中毒并失活;油品中的含硫、含氮化合物在燃烧时会增加so
x
、no
x
及颗粒物排放,从而污染环境;碱性含氮化合物和苯并噻吩类硫化物会严重影响柴油的氧化安定性及色度。因此,在油品加工过程中,油品中含硫、含氮化合物的脱除是柴油加工的重点工作之一,为了更好的促进加氢脱硫、加氢脱氮催化剂的改进及工艺技术的升级,对柴油原料和产品中的含硫、含氮化合物进行研究,获得其分子组成数据尤为重要。
3.含硫、含氮化合物在柴油馏分中的含量较低,仅依靠仪器无法在众多烃类化合物的干扰下得到精准的单体信息,这就需要借助预处理手段将其从油品中分离出来后,再进行下一步的分子识别工作。固相萃取法具备操作步骤简单、溶剂用量少、操作条件易优化等优点,常作为一种预处理手段用于柴油馏分中含硫及含氮化合物的分离。孔翠萍等[孔翠萍,等.全二维气相色谱分析柴油中的硫化物分布[j].石油炼制与化工,2016,47(9):106-110]采用固相萃取法分离了直馏柴油中的含硫化合物。方法中包含两个部分,先将柴油馏分分离为饱和烃和芳香烃组分,再采用负载氯化钯的硅胶柱将芳香烃组分分离为芳烃化合物和含硫化合物。该分离方法可得到较为纯净的含硫化合物,但是存在分离步骤复杂,溶剂用量大的缺点。
[0004]
固相萃取法在柴油含氮化合物的分离工作中已有较多的应用,张月琴等[张月琴.催化裂化柴油中含氮化合物类型分布[d].,2013,44(5)87-91.]采用固相萃取法分离了柴油含氮化合物,先采用中性硅胶柱分离富集含氮化合物,又进一步使用酸改性硅胶柱将含氮化合物分离为碱性含氮化合物和中性含氮化合物,取得了较好的分离效果。wiwel等[wiwel p,knudsen k,zeuthen p,et al.assessing compositional changes of nitrogen compounds during hydrotreating of typical diesel range gas oils using a novel preconcentration technique coupled with gas chromatography and atomic emission detection[j].industrial&engineering chemistry research.2000,39(2):533-540.]以纯硅胶作为固定相,采用固相萃取法分离富集了科威特柴油与轻循环油混合物中的含氮化合物,含氮化合物的回收率可高达99%。史得军等[史得军,陈菲,梁迎春,等.固相萃取/气相色谱-质谱分析催化裂化柴油中的含氮化合物[j].分析测试学报,2018,37(12):1490-1494.]采用硅胶和中性氧化铝混合物作为固定相建立了分离柴油含氮化合物的方法,其回收率高达99.5%,且具有溶剂用量少,分离周期短的优势。
[0005]
通过文献调研可知,固相萃取法已较多的应用于柴油含硫、含氮化合物的分离过
程中,但是诸多的预处理手段均为只能分离含硫化合物或含氮化合物中的其中一种,暂未有可以同时分离柴油中含硫及含氮化合物的方法报道。含硫、含氮化合物的分布和含量是油品加工过程中的常用数据,如果能够将两类化合物的分离工作集中在一个固相萃取方法中,可以大幅度简化分离步骤,提高工作效率。
技术实现要素:
[0006]
本发明目的是提供一种分离柴油中含氮化合物和含硫化合物的固相萃取方法,其中包括固相萃取柱和固相萃取流程。可实现在一次分离过程中同时富集柴油馏分中的含氮化合物和含硫化合物,并且能够解决含硫化合物或含氮化合物分离过程复杂、耗时长、溶剂用量大等问题。
[0007]
本发明所述分离柴油馏分中含氮化合物和含硫化合物的固相萃取方法主要步骤为:
[0008]
步骤a、取固相萃取柱a,采用第一洗脱溶剂润湿固相萃取柱a,将柴油样品加入固相萃取柱a上层,采用第一洗脱溶剂冲洗,得到饱和烃组分;
[0009]
步骤b、取固相萃取柱b,采用第二洗脱溶剂润湿固相萃取柱b,并串联固相萃取柱a和b,固相萃取柱a在上,采用第二洗脱溶剂冲洗固相萃取柱a和b,得到芳烃组分;
[0010]
步骤c、将固相萃取柱a和b分开,采用第三洗脱溶剂冲洗固相萃取柱a,得到含氮化合物组分;
[0011]
步骤d、采用第四洗脱溶剂冲洗固相萃取柱b,得到含硫化合物组分。
[0012]
本发明所述的固相萃取柱a中装填有固定相,所述固定相为硅胶和氧化铝的混合物,其中硅胶含量为5-50wt%,优选5-30wt%。
[0013]
本发明所述的固相萃取柱b中装填有固定相,所述固定相为负载氯化钯的硅胶和氧化铝混合物,其中负载氯化钯的硅胶含量为0-20wt%,优选0-10wt%;钯的负载量为3-8wt%,优选4-6wt%。
[0014]
本发明所述的固相萃取柱b的固定相进行钯负载时需要在温度为80-120℃下搅拌20-30min。
[0015]
本发明所述的固相萃取柱a和b制备中所用硅胶的比表面积为300-600m2/g,优选450-600m2/g,孔体积为0.2-0.9ml/g,优选0.3-0.6ml/g,平均孔径为2-6nm;氧化铝比表面积为100-300m2/g,优选150-250m2/g,孔体积为0.1-0.5ml/g,优选0.2-0.4ml/g,平均孔径3-5nm。
[0016]
本发明所述的固相萃取柱a固定相制备过程中硅胶活化条件为100-150℃干燥3-6h,优选120-140℃干燥4-5h,氧化铝活化条件为300-600℃焙烧1-5h,优选350-500℃焙烧2-3h。
[0017]
本发明所述固相萃取柱b固定相的制备为:硅胶100-150℃干燥3-6h,得到活化硅胶,氧化铝300-600℃干燥2-6h,得到活化氧化铝。将活化好的硅胶和氧化铝分别与氯化钯溶液以1:1.0-1.5的体积比均匀混合,80-120℃加热搅拌20-30min,直至呈现出黏糊状,持续的加热和搅拌过程可以使pdcl2与硅胶或氧化铝结合的更加紧密,最大程度避免在后续溶剂洗脱过程中钯流失的问题。避光浸渍20-30h后,100-150℃条件下干燥负载氯化钯的硅胶3-6h,优选条件为120-140℃干燥4-5h;300-500℃条件下焙烧负载氯化钯的氧化铝1-4h,
优选条件为350-500℃焙烧2-3h,在干燥器中冷却至室温,按照一定质量比均匀混合pd-sio2和pd-al2o3。
[0018]
本发明固相萃取流程中所述第一洗脱溶剂选自正戊烷、正己烷、正庚烷中的至少一种;第二洗脱溶剂为a和b的混合溶液,a选自正戊烷、正己烷、正庚烷中的至少一种,b选自二氯甲烷、氯仿、乙醚、四氯化碳、甲苯中的至少一种,第二洗脱溶剂中a和b的体积比为2-7:1;第三洗脱溶剂为c和二氯甲烷的混合溶液,c选自丙酮、甲醇、乙醇中的至少一种,第三洗脱溶剂中c与二氯甲烷的体积比为0.1-0.6:1;第四洗脱溶剂为d和二氯甲烷的混合溶液,d选自乙腈、丙酮、异丙醇、乙醇中的至少一种,第四洗脱溶剂中d与二氯甲烷的体积比为0.15-1:1。
[0019]
本发明所述冲洗饱和烃组分时所用第一洗脱溶剂与柴油样品的体积比为1-8:1。冲洗芳烃组分时所用第二洗脱溶剂与柴油样品体积比为3-30:1;冲洗含氮化合物时所用第三洗脱溶剂与柴油样品体积比为2-25:1;洗脱含硫化合物组分时所用第四洗脱溶剂与柴油样品体积比为2-20:1。
[0020]
本发明所述的柴油样品为直馏柴油、催化裂化柴油、加氢柴油、焦化柴油、成品柴油。所处理柴油样品量与固相萃取柱a的固定相填料质量比为1:1-6。
[0021]
本发明所述的方法分离柴油馏分后可得到饱和烃、芳烃、含氮化合物及含硫化合物溶液,经溶剂挥发浓缩后即可得到相应的组分,优先选用旋转蒸发、氮气吹扫进行溶剂挥发,浓缩至0.2-0.5ml即可,采用气相色谱-质谱(gc-ms)进行定性分析工作,气相色谱-氮化学发光检测器(gc-ncd)及气相色谱
–
硫化学发光检测器(gc-scd)结合内标法进行定量分析工作,含氮化合物内标物优选吡咯,含硫化合物内标物优选3-甲基噻吩。
[0022]
本发明提供一种分离柴油中含氮化合物和含硫化合物的方法。组合使用两根固定相完全不同的固相萃取柱,根据不同类型化合物极性不同的特点,通过调整洗脱顺序、洗脱溶剂的极性及用量,可以实现在一次固相萃取方法中同时富集柴油中的含氮化合物和含硫化合物两个组分。该方法大大简化了分离步骤、提高了分离效率,并且溶剂用量少、操作简单快捷,分离所得含氮化合物组分和含硫化合物组分可满足后续仪器分析需求。
附图说明
[0023]
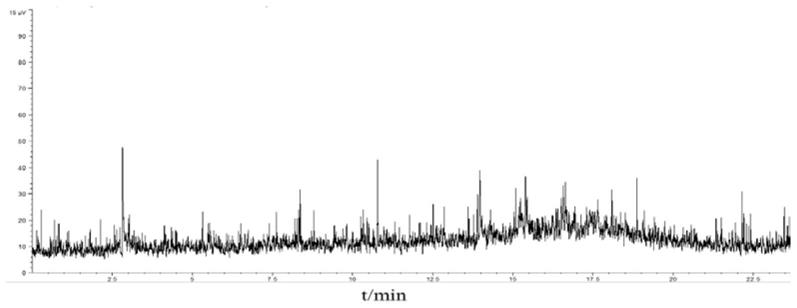
图1为本发明实施例1的方法中催化柴油芳烃组分gc-scd色谱图;
[0024]
图2为本发明实施例1的方法中催化柴油含氮化合物组分gc-ncd色谱图;
[0025]
图3为本发明实施例1的方法中催化柴油含氮化合物组分gc/ms总离子流色谱图
[0026]
图4为本发明实施例1的方法中催化柴油含硫化合物组分gc-scd色谱图;
[0027]
图5为本发明实施例1的方法中催化柴油含硫化合物gc/ms总离子流色谱图。
具体实施方式
[0028]
下面将对本发明的实施例中的技术方案进行详细的说明,但如下实施例仅适用以理解本发明,而不能限制本发明,本发明可以由权利要求限定和覆盖的多种不同方式实施。
[0029]
实施例中所用的硅胶为国药集团化学试剂有限公司生产的层析用硅胶,颗粒度≥70.0%,干燥失量≤6.0%,比表面积为511.9m2/g,孔体积为0.468ml/g。氧化铝为国药集团化学试剂有限公司生产的层析用中性氧化铝,颗粒度≥75.0%,灼烧失重≤8.0%,比表面
积为177.8m2/g,孔体积为0.255ml/g。
[0030]
实施例中制备固相萃取柱a固定相步骤为将硅胶于140℃干燥4h,得到活化好的硅胶,放入干燥器备用。将中性氧化铝于450℃焙烧3h,得到活化好的氧化铝,按照一定质量比均匀混合硅胶氧化铝,得到柱a的固定相。
[0031]
实施例中制备固相萃取柱b固定相步骤为将硅胶于150℃干燥2h,中性氧化铝于500℃焙烧2h,称取两份1.2g氯化钯分别溶解于25ml热水中,称取活化好硅胶和氧化铝各20g,分别加入氯化钯溶液中,边加热边搅拌直至黏糊状;避光浸渍24h后,硅胶150℃干燥5h,得到pd-sio2;氧化铝400℃焙烧2.5h,得到pd-al2o3。按照一定质量比均匀混合pd-sio2和pd-al2o3固定相,得到萃取柱b的固定相。
[0032]
实施例中所用的gc-ms仪器型号为7890agc-5975ms,带fid检测器。gc条件:hp-pona毛细管色谱柱,50m
×
0.2mm
×
0.5μm;程序升温为初温60℃,保持2min后以10℃/min升至300℃,保持8min;载气为高纯氦,恒流操作0.8ml/min;进样口温度300℃,分流比30:1,进样量1μl。ms条件:ei电离源(70ev),离子源温度230℃,四级杆温度130℃,全扫描质量范围30-500u,接口温度300℃,溶剂延迟4min。fid条件:检测器温度350℃,空气流量为300ml/min,氢气流量为30ml/min。gc-ncd型号为7890a gc-255ncd。gc条件:hp-pona毛细管色谱柱,50m
×
0.2mm
×
0.5μm;程序升温初温120℃,升温速率4℃/min,终温270℃,保持10min;载气为高纯氦,恒流操作0.8ml/min;进样口温度280℃,分流比30:1,进样量1μl。ncd条件:燃烧器温度900℃;氢气流速5ml/min;氧气流速10ml/min。gc-scd型号为7890agc-355scd。gc条件:hp-pona毛细管色谱柱,50m
×
0.2mm
×
0.5μm;程序升温初温120℃,以4℃/min升至270℃,保持10min;载气为高纯氦,恒流操作0.8ml/min;进样口温度280℃,分流比30:1,进样量1μl。scd条件:燃烧器温度800℃;空气流速60ml/min;氢气流速40ml/min。
[0033]
实施例1
[0034]
固相萃取柱a中装填3g的5wt%sio
2-al2o3固定相;柱b固定相中装填3.5g的pd-al2o3固定相,钯负载量为8wt%。
[0035]
用2ml第一洗脱溶剂(正己烷)润湿柱a,称取0.5g(约0.6ml)催化柴油于柱a上层,用3ml第一洗脱溶剂(正己烷)冲洗柱a得到饱和烃组分;用2ml第二洗脱溶剂(正己烷-二氯甲烷体积比4:1)润湿柱b,将a、b两柱串联,a柱在上层,用14ml第二洗脱溶剂(正己烷-二氯甲烷体积比4:1)冲洗柱a和柱b,得到芳烃组分;分开a、b两柱,用8ml第三洗脱溶剂(乙醇-二氯甲烷体积比为1:2)冲洗柱a,得到含氮化合物组分。用10ml第四洗脱溶剂(丙酮-二氯甲烷体积比1:3)冲洗柱b,得到含硫化合物组分。采用氮气吹扫方式将各组分溶剂浓缩至0.5ml备测。分离过程总计用时20min。
[0036]
采用gc-ncd及gc-scd分析饱和烃及芳烃组分,结果表明其中含硫及含氮化合物的信号响应值均较低。采用gc-ncd及gc/ms(fid)分析含氮化合物组分,gc-ncd色谱图及总离子流色谱图见图2及图3,结果表明含氮化合物组分中主要为含氮化合物。结合质谱定性并采用面积归一化法定量,得到其中杂质含量约为2.9%。采用gc-scd及gc/ms(fid)分析含硫化合物组分,gc-scd色谱图及总离子流色谱图见图4及图5,结果表明含硫化合物组分中主要为含硫化合物,结合质谱定性并采用面积归一化法定量,得到其中杂质含量约为6.1wt%,杂质的存在并不影响含硫化合物定性分析。
[0037]
以咔唑和二苯并噻吩的加标回收率考察本发明分离柴油中含氮、含硫化合物的回
收率。取两份0.5g的柴油样品,其中一份加入一定量的咔唑和二苯并噻吩,两份样品按照上述分离方法分离得到组分3和组分4,氮气吹扫浓缩至0.5ml左右,向组分3加入30μl浓度为4067μg/ml的吡咯溶液作为内标,采用gc-ncd进行分析;组分4中加入25μl浓度为4010μg/ml的3-甲基噻吩溶液作为内标,采用gc-scd进行分析。内标法计算加标样品中咔唑及二苯并噻吩的含量减去未加标样品中咔唑及二苯并噻吩含量,其差值同加入的咔唑及二苯并噻吩标样含量相比即为其加标回收率,其值分别为97.5%、90.6%。
[0038]
综上所述,本发明的分离方法分离步骤简单、组分间交叉较小、回收率较高,分离效果良好。
[0039]
对比例1
[0040]
固相萃取柱a中装填3g的40wt%sio
2-al2o3固定相用于分离柴油中含氮化合物;固相萃取柱b装填3g的pd-sio2固定相用于分离含硫化合物;固相萃取柱c中装填3g硅胶用于分离饱和烃和芳香烃组分。pb-sio2制备过程为140℃干燥4h得到活化硅胶,取一定量氯化钯溶解于蒸馏水中,并倒入一定量活化硅胶,浸渍24h,以150℃干燥3h,制备得到钯负载量为6wt%的pd-sio2。所用硅胶和氧化铝的活化条件与该实例中一致。
[0041]
采用固相萃取柱a分离柴油中的含氮化合物,用2ml正己烷-二氯甲烷混合液(体积比3:1)润湿柱a,称取0.5g催化柴油置于柱a上层,用12ml正己烷-二氯甲烷混合液(体积比3:1)冲洗柱a得到饱和烃和芳香烃混合组分;用12ml丙酮-二氯甲烷混合液(体积比1:4)冲洗柱a得到含氮化合物组分。采用与实施例1中同样的方法测定含氮化合物组分中杂质含量,其值约为2.8%。咔唑的加标回收率为95.8%。
[0042]
组合使用固相萃取柱b和c分离柴油中的含硫化合物。先用2ml正己烷润湿柱c,称取0.5g催化柴油置于柱c上层,用3ml正己烷冲洗柱a得到饱和烃组分;采用14ml二氯甲烷冲洗柱c,得到芳香烃组分。氮气吹扫方式蒸干芳香烃组分中的溶剂。用2ml正己烷-二氯甲烷体积比9:1润湿柱b,将芳香烃组分加入柱b上层,用14ml正己烷-二氯甲烷混合液(体积比9:1)冲洗柱b,得到芳烃组分;用12ml异丙醇-二氯甲烷混合液(体积比3:17)冲洗固相萃取柱b,得到含硫化合物组分。其中分离得到的含硫芳烃组分为深褐色,说明柱b上的钯大量流失,需要加入一定量的二乙胺后再进行溶剂浓缩。采用与实施例1中同样的方法测定含硫芳烃中的杂质,其值为8.7wt%,二苯并噻吩的加标回收率为83.2%。
[0043]
以上方法分离柴油中的含氮及含硫化合物用时长达60min,与实施例1相比,存在耗时长,溶剂用量大,含硫化合物回收率低等问题。
[0044]
实施例2
[0045]
固相萃取柱a中装填3g的30wt%sio
2-al2o3固定相;柱b固定相中装填3.5g的10wt%pd-sio2/pd-al2o3固定相,钯负载量为5wt%。
[0046]
用2ml第一洗脱溶剂(正戊烷)润湿柱a,称取1g(约1.2ml)直馏柴油于柱a上层,用3ml第一洗脱溶剂(正戊烷)冲洗柱a得到饱和烃组分;用2ml第二洗脱溶剂(正戊烷-二氯甲烷体积比4:1)润湿柱b,将a、b两柱串联,a柱在上层,用16ml第二洗脱溶剂(正戊烷-二氯甲烷体积比4:1)冲洗柱a和柱b,得到芳烃组分;分开a、b两柱,用8ml第三洗脱溶剂(丙酮-二氯甲烷体积比为1:3)冲洗柱a,得到含氮化合物组分。用10ml第四洗脱溶剂(乙腈-二氯甲烷体积比1:3)冲洗柱b,得到含硫化合物组分。
[0047]
采用与实施例1一致的方法浓缩各组分并进行分析,结果表明含氮化合物中杂质
含量约为2.8wt%;含硫化合物组分中杂质含量约为4.1wt%。
[0048]
咔唑的加标回收率为102.5%,二苯并噻吩加标回收率为90.4%。
[0049]
实施例3
[0050]
固相萃取柱a中装填4g的50wt%sio
2-al2o3固定相;柱b固定相中装填3.5g的20wt%pd-sio2/pd-al2o3固定相,钯负载量为6wt%。
[0051]
用2ml第一洗脱溶剂(正己烷)润湿柱a,称取3g(约3.7ml)加氢柴油于柱a上层,用4ml第一洗脱溶剂(正己烷)冲洗柱a得到饱和烃组分;用2ml第二洗脱溶剂(正己烷-二氯甲烷体积比4:1)润湿柱b,将a、b两柱串联,a柱在上层,用14ml第二洗脱溶剂(正己烷-二氯甲烷体积比4:1)冲洗柱a和柱b,得到芳烃组分;分开a、b两柱,用8ml第三洗脱溶剂(甲醇-二氯甲烷体积比为1:9)冲洗柱a,得到含氮化合物组分。用10ml第四洗脱溶剂(异丙醇-二氯甲烷体积比1:3)冲洗柱b,得到含硫化合物组分。
[0052]
采用与实施例1一致的方法浓缩各组分并进行分析,结果表明含氮化合物中杂质含量约为3.0wt%;含硫化合物组分中杂质含量约为4.0wt%。
[0053]
咔唑的加标回收率为97.5%,二苯并噻吩加标回收率为91.9%。
[0054]
实施例4
[0055]
固相萃取柱a中装填4g的50wt%sio
2-al2o3固定相;柱b固定相中装填5.0g的10wt%pd-sio2/pd-al2o3固定相,钯负载量为6wt%。
[0056]
用2ml第一洗脱溶剂(正庚烷)润湿柱a,称取2g(约2.5ml)催化柴油于柱a上层,用4ml第一洗脱溶剂(正庚烷)冲洗柱a得到饱和烃组分;用2ml第二洗脱溶剂(正庚烷-二氯甲烷体积比3:1)润湿柱b,将a、b两柱串联,a柱在上层,用12ml第二洗脱溶剂(正庚烷-二氯甲烷体积比3:1)冲洗柱a和柱b,得到芳烃组分;分开a、b两柱,用12ml第三洗脱溶剂(甲醇-二氯甲烷体积比为1:9)冲洗柱a,得到含氮化合物组分。用12ml第四洗脱溶剂(异丙醇-二氯甲烷体积比1:3)冲洗柱b,得到含硫化合物组分。
[0057]
采用与实施例1一致的方法浓缩各组分并进行分析,结果表明含氮化合物中杂质含量约为4.6wt%;含硫化合物组分中杂质含量约为5.1wt%。
[0058]
咔唑的加标回收率为90.2%,二苯并噻吩加标回收率为88.9%。
[0059]
实施例5
[0060]
固相萃取柱a中装填4g的50wt%sio
2-al2o3固定相;柱b固定相中装填5.0g的10wt%pd-sio2/pd-al2o3固定相,钯负载量为3wt%。
[0061]
用2ml第一洗脱溶剂(正已烷)润湿柱a,称取1g(约1.2ml)催化柴油于柱a上层,用3ml第一洗脱溶剂(正己烷)冲洗柱a得到饱和烃组分;用2ml第二洗脱溶剂(正己烷-甲苯体积比17:3)润湿柱b,将a、b两柱串联,a柱在上层,用12ml第二洗脱溶剂(正己烷-甲苯体积比17:3)冲洗柱a和柱b,得到芳烃组分;分开a、b两柱,用12ml第三洗脱溶剂(丙酮-二氯甲烷体积比为1:4)冲洗柱a,得到含氮化合物组分。用12ml第四洗脱溶剂(丙酮-二氯甲烷体积比1:1)冲洗柱b,得到含硫化合物组分。
[0062]
采用与实施例1一致的方法浓缩各组分并进行分析,结果表明含氮化合物中杂质含量约为5.1wt%;含硫化合物组分中杂质含量约为6.2wt%。
[0063]
咔唑的加标回收率为92.3%,二苯并噻吩加标回收率为91.2%。
[0064]
实施例6
[0065]
固相萃取柱a中装填5g的50wt%sio
2-al2o3固定相;柱b固定相中装填4.5g的5wt%pd-sio2/pd-al2o3固定相,钯负载量为6wt%。
[0066]
用2ml第一洗脱溶剂(正己烷)润湿柱a,称取2g(约2.5ml)催化柴油于柱a上层,用5ml第一洗脱溶剂(正己烷)冲洗柱a得到饱和烃组分;用2ml第二洗脱溶剂(正己烷-四氯化碳体积比3:1)润湿柱b,将a、b两柱串联,a柱在上层,用12ml第二洗脱溶剂(正己烷-四氯化碳体积比3:1)冲洗柱a和柱b,得到芳烃组分;分开a、b两柱,用12ml第三洗脱溶剂(甲醇-二氯甲烷体积比为1:9)冲洗柱a,得到含氮化合物组分。用12ml第四洗脱溶剂(乙醇-二氯甲烷体积比3:17)冲洗柱b,得到含硫化合物组分。
[0067]
采用与实施例1一致的方法浓缩各组分并进行分析,结果表明含氮化合物中杂质含量约为3.9wt%;含硫化合物组分中杂质含量约为5.4wt%。
[0068]
咔唑的加标回收率为93.6%,二苯并噻吩加标回收率为94.2%。