1.本发明属于药物合成技术领域,具体涉及一种甲苯达唑的制备方法。
背景技术:
2.甲苯达唑为白色、类白色或微黄色结晶性粉末,属于苯并咪唑氨基甲酸酯类药物,是在八十年代合成并投入临床的驱虫药,对蛔虫、钩虫、蛲虫、鞭虫、绦虫和粪类圆线虫等肠道蠕虫均有效,具有广谱、高效、安全性高的特点。
3.甲苯达唑合成方式主要以对氯苯甲酸为原料,通过硝化、傅克、氨化、还原、缩合一系列步骤后得到甲苯达唑,该流程反应步骤多,收率低,原子利用率只能达到49.77%,且涉及到硝化、氨化与还原等危险工艺,安全性差,审批困难,工业化可行性差。
4.专利cn106083622b介绍了在钯-镍等金属负载催化剂存在的条件下,以四氢吡咯等为氢供体,通过转移氢化还原的方式还原4-氨基-3-硝基二苯甲酮。该工艺尽管降低了催化加氢的反应温度与压力,但催化剂浸渍制备时间长,操作复杂,工业化难度大,并且使用水合肼等还原剂进行催化剂活化,成本较高。
5.专利cn109467512b中使用氨水代替液氨进行3-硝基-4-氯-二苯甲酮的氨化,并通过钯碳催化加氢制备3,4-二氨基二苯甲酮。该法尽管降低了杂质含量,但是反应条件严苛,对设备具有较高要求,而且反应时间较长,处理步骤较多,操作复杂,仍然不能令人满意。
6.而且,上述专利公开的制备方法都以改进现有工艺为主,并未从根本上解决反应流程长,工艺危险等问题。
技术实现要素:
7.本发明提供一种甲苯达唑的制备方法,该制备方法以三氯甲苯为原料,通过催化水解得到苯甲酰氯,苯甲酰氯与多菌灵进行傅克反应得到甲苯达唑,该方法摒弃了传统的先傅克后成环的方法,以多菌灵为反应物,直接合成甲苯达唑,总收率高,相对于传统工艺,具有流程简单,条件温和,原子利用率高的特点。
8.为解决以上技术问题,本发明采取的技术方案如下:一种甲苯达唑的制备方法,包括酰化反应和傅克反应。
9.所述酰化反应,将三氯甲苯升温至50-90℃后滴加氯化锌水溶液,控制氯化锌水溶液的滴加时间为50-70min,滴加期间保持温度在50-90℃,滴加结束继续保温反应50-80min,待反应完毕,减压蒸馏得到苯甲酰氯。
10.其中,氯化锌水溶液中的氯化锌与三氯甲苯的摩尔比为0.1-0.2:1。
11.其中,氯化锌水溶液中的水与三氯甲苯的摩尔比为1.05-1.15:1。
12.所述傅克反应,将多菌灵、溶剂、无水氯化铝混合搅拌后降温至10-20℃,然后滴加苯甲酰氯,50-70min滴完,然后继续搅拌保温反应2.5-3.5h,反应完毕,减压蒸馏回收乙酸后得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑。
13.其中,无水氯化铝与苯甲酰氯的摩尔比为1.1-1.3:1。
14.其中,多菌灵与苯甲酰氯的摩尔比为1-1.1:1。
15.所述溶剂为甲酸或乙酸中的一种。
16.所述溶剂与苯甲酰氯的摩尔比为6.8-8.9:1。
17.与现有技术相比,本发明的有益效果为:(1)本发明的甲苯达唑的制备方法,能够显著缩短反应流程,减少中间损失,无需经过高压加氢与氨化,反应条件相对温和,工业化难度低,具有很强的发展空间与竞争能力;(2)本发明的甲苯达唑的制备方法,原子利用率能达到72.97%,与现有的以对氯苯甲酸为原料的制备方法相比,原子利用率提高了23.2%;(3)本发明的甲苯达唑的制备方法,制备的中间体苯甲酰氯的摩尔收率为85.44%-96.47%,气相检测纯度为100%;(4)本发明的甲苯达唑的制备方法,制备的产品甲苯达唑的摩尔收率能达到89.69%-94.51%,液相检测纯度为99.1%-99.8%,合计总收率为79.72%-90.04%。
附图说明
18.图1为制备甲苯达唑的反应过程;图2为标准品溶液的液相色谱图;图3为实施例1制备的甲苯达唑溶液的液相色谱图。
具体实施方式
19.为了对本发明的技术特征、目的和效果有更加清楚的理解,现说明本发明的具体实施方式。
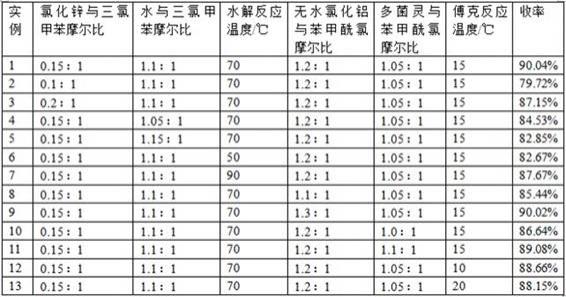
20.实施例1如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,然后取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.61g(0.1964mol)苯甲酰氯,摩尔收率96.00%,气相检测纯度为100%。
21.2.傅克反应:取39.44g(0.2063mol)多菌灵、80g(1.3322mol)乙酸、31.43g(0.2357mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑54.62g,摩尔收率93.79%,液相检测纯度为99.6%,合计总收率90.04%。
22.所述摩尔收率的计算公式为:n(甲苯达唑)/n(苯甲酰氯)所述总收率的计算公式为:n(甲苯达唑)/n(三氯甲苯)采用相同的溶解方法溶解标准品和本实施例制备的产品甲苯达唑后得到标准品溶液和甲苯达唑溶液,然后分别对标准品溶液和甲苯达唑溶液进行液相色谱分析,标准品溶液的液相色谱图如图2所示,甲苯达唑溶液的液相色谱图如图3所示。
23.实施例2
如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取2.79g(0.0205mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,然后取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到24.58g(0.1748mol)苯甲酰氯,摩尔收率85.44%,气相检测纯度为100%。
24.2.傅克反应:取35.10g(0.1836mol)多菌灵、80g(1.3322mol)乙酸、27.97g(0.2098mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑48.46g,摩尔收率93.31%,液相检测纯度为99.4%,合计总收率79.72%。
25.实施例3如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取5.58g(0.0409mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,然后取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到26.68g(0.1898mol)苯甲酰氯,摩尔收率92.74%,气相检测纯度为100%。
26.2.傅克反应:取38.10g(0.1993mol)多菌灵、80g(1.3322mol)乙酸、30.36g(0.2277mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑52.87g,摩尔收率93.97%,液相检测纯度为99.6%,合计总收率87.15%。
27.实施例4如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和3.87g(0.2149mol)水,搅拌溶解后得到氯化锌水溶液,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到26.22g(0.1865mol)苯甲酰氯,摩尔收率91.15%,气相检测纯度为100%。
28.2.傅克反应:取37.45g(0.1958mol)多菌灵、80g(1.3322mol)乙酸、29.84g(0.2238mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑51.54g,摩尔收率92.74%,液相检测纯度为99.1%,合计总收率84.53%。
29.实施例5如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.24g(0.2353mol)水,搅拌溶解后得到氯化锌水溶液,,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到25.35g(0.1804mol)苯甲酰氯,摩尔收率88.14%,气相检测纯度为100%。
30.2.傅克反应:取36.21g(0.1894mol)多菌灵、80g(1.3322mol)乙酸、28.86g
(0.2164mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑50.21g,摩尔收率94.00%,液相检测纯度为99.7%,合计总收率82.85%。
31.实施例6如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,然后取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至50℃,开始滴加氯化锌水溶液,控制滴加时间为50min,滴加期间保持温度在50℃,滴加结束继续保温反应50min,待反应完毕,减压蒸馏得到25.75g(0.1832mol)苯甲酰氯,摩尔收率89.51%,气相检测纯度为100%。
32.2.傅克反应:取36.77g(0.1923mol)多菌灵、80g(1.3322mol)乙酸、29.31g(0.2198mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑50.45g,摩尔收率92.74%,液相检测纯度为99.4%,合计总收率82.67%。
33.实施例7如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,然后取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至90℃,开始滴加氯化锌水溶液,控制滴加时间为70min,滴加期间保持温度在90℃,滴加结束继续保温反应80min,待反应完毕,减压蒸馏得到27.52g(0.1958mol)苯甲酰氯,摩尔收率95.68%,气相检测纯度为100%。
34.2.傅克反应:取39.31g(0.2056mol)多菌灵、80g(1.3322mol)乙酸、31.33g(0.2349mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑53.29g,摩尔收率91.63%,液相检测纯度为99.4%,合计总收率87.67%。
35.实施例8如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.40g(0.1949mol)苯甲酰氯,摩尔收率95.26%,气相检测纯度为100%。
36.2.傅克反应:取39.14g(0.2047mol)多菌灵、80g(1.7391mol)甲酸、28.59g(0.2144mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收甲酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑51.90g,摩尔收率89.69%,液相检测纯度为99.5%,合计总收率85.44%。
37.实施例9如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌
水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.40g(0.1949mol)苯甲酰氯,摩尔收率95.25%,气相检测纯度为100%。
38.2. 傅克反应:取39.13g(0.2046mol)多菌灵、80g(1.3322mol)乙酸、33.78g(0.2534mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑54.50g,摩尔收率94.51%,液相检测纯度为99.8%,合计总收率90.02%。
39.实施例10如图1所示,一种甲苯达唑的制备方法,具体为:1. 酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.45g(0.1953mol)苯甲酰氯,摩尔收率95.42%,气相检测纯度为100%。
40.2.傅克反应:取37.33g(0.1953mol)多菌灵、80g(1.3322mol)乙酸、31.24g(0.2343mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑52.63g,摩尔收率90.79%,液相检测纯度为99.5%,合计总收率86.64%。
41.实施例11如图1所示,一种甲苯达唑的制备方法,具体为:1. 酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,然后取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.50g(0.1956mol)苯甲酰氯,摩尔收率95.59%,气相检测纯度为100%。
42.2. 傅克反应:取41.14g(0.2152mol)多菌灵、80g(1.3322mol)乙酸、31.30g(0.2347mol)无水氯化铝至烧瓶中,搅拌降温至15℃,滴加步骤1制备的苯甲酰氯,1h滴完,然后继续搅拌保温3h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑54.26g,摩尔收率93.19%,液相检测纯度为99.2%,合计总收率89.08%。
43.实施例12如图1所示,一种甲苯达唑的制备方法,具体为:1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.74g(0.1973mol)苯甲酰氯,摩尔收率96.44%,气相检测纯度为100%。
44.2.傅克反应:取39.62g(0.2072mol)多菌灵、80g(1.3322mol)乙酸、31.58g(0.2368mol)无水氯化铝至烧瓶中,搅拌降温至10℃,滴加步骤1制备的苯甲酰氯,50min滴完,然后继续搅拌保温2.5h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑53.84g,摩尔收率91.93%,液相检测纯度为99.5%,合计总收率88.66%。
45.实施例13如图1所示,一种甲苯达唑的制备方法,具体为:
1.酰化反应:取4.18g(0.0307mol)氯化锌和4.05g(0.2251mol)水,搅拌溶解后得到氯化锌水溶液,取40g(0.2046mol)三氯甲苯至四口烧瓶中,升温至70℃,开始滴加氯化锌水溶液,控制滴加时间为1h,滴加期间保持温度在70℃,滴加结束继续保温反应1h,待反应完毕,减压蒸馏得到27.75g(0.1973mol)苯甲酰氯,摩尔收率96.47%,气相检测纯度为100%。
46.2. 傅克反应:取39.63g(0.2073mol)多菌灵、80g(1.3322mol)乙酸、31.59g(0.2369mol)无水氯化铝至烧瓶中,搅拌降温至20℃,滴加步骤1制备的苯甲酰氯,70min滴完,然后继续搅拌保温3.5h。反应完毕,减压蒸馏回收乙酸得到粗品,经水洗、乙酸重结晶,得到产品甲苯达唑53.47g,摩尔收率91.37%,液相检测纯度为99.6%,合计总收率88.15%。
47.实施例2-13中摩尔收率和总收率的计算公式与实施例1相同。
48.实施例1-13数据汇总表除非另有说明,本发明中所采用的百分数均为质量百分数。
49.最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。