一种用于二胺检测的aie柱芳烃荧光探针及其制法与应用
技术领域
1.本发明属于有机合成以及分析检测技术领域,特别涉及一种用于二胺检测的aie柱芳烃荧光探针及其制法与应用。
背景技术:
2.烷基二胺是一类重要的工业原料,尤其是具有较长烷基链的二胺(c≥8)在精细化工领域具有十分广泛的应用(例如1,10-二氨基癸烷是合成尼龙-1010的重要中间体)。近年来,研究人员合成了大量可以检测烷基二胺的荧光传感器,但其中也有一个很明显的问题,那就是这些荧光传感器对具有不同烷基链长度的二胺的选择性不够高。(strutt,n.;forgan,r.;spruell,j.;botros,y.;stoddart,j.,monofunctionalized pillar[5]arene as a host for alkanediamines.j.am.chem.soc.2011,133,5668-5671.)。这是因为所有的烷基二胺均属于同系物,它们的化学性质十分相似,主要的不同就表现在亚甲基的数量上,从而导致很难将它们区分开。因此研发一种可以选择性地检测特定烷基链长度的二胺的荧光探针成为迫切需求。
[0003]
柱[5]芳烃具有结构刚性、易于功能化和良好的主客体性能等特点。目前,柱[5]芳烃功能化研究最广泛的策略之一是将其与聚集诱导发光(aie)基团共价连接。大量具有aie性质的柱[5]芳烃已经成功被用于气体吸附、离子或小分子检测、染料敏化太阳能电池和人工光收集等领域(li,j.;wang,j.;li,h.;song,n.;wang,d.;tang,b.,supramolecular materials based on aie luminogens(aiegens):construction and applications.chem.soc.rev.2020,49,1144-1172.)。因此,利用具有aie性质的柱[5]芳烃来实现对客体的精准识别是一种具有较大应用前景的策略。
[0004]
喹啉-丙二腈单元是一种良好的aie构建骨架,在柱[5]芳烃中引入该基团可以很好地提升柱芳烃的荧光性质。其次,通过在柱芳烃的两端引入两个特定的锚固基团可以使主、客体在结合过程中不只有柱芳烃空腔与客体之间的相互作用,两个锚固基团还能提供两个额外的相互作用力来提高两者的结合能力。通过多个结合位点以及多种作用力协同作用便可实现对特定二胺的选择性检测。
技术实现要素:
[0005]
本发明的目的是针对烷基二胺荧光探针种类稀少、选择性差以及灵敏度低的不足,设计合成了一种用于二胺检测的aie柱芳烃荧光探针。
[0006]
本发明的目的通过如下技术方案实现。
[0007]
本发明提供的一种用于二胺检测的aie柱芳烃荧光探针分子,其结构式为:
[0008][0009]
本发明提供的一种合成上述aie柱芳烃荧光探针分子的方法,包括以下步骤:
[0010]
(1)以化合物1为起始原料,与2-噻吩硼酸在催化剂作用下发生suzuki偶联反应生成化合物2;
[0011]
(2)化合物2与n,n-二甲基甲酰胺在催化剂作用下发生甲酰化反应生成化合物3;
[0012]
(3)化合物3与四乙基氢氧化铵发生水解反应生成化合物4;
[0013]
(4)化合物4与溴乙酸乙酯发生williamson反应生成化合物5;
[0014]
(5)以2-甲基喹啉为起始原料,与溴乙酸乙酯发生取代反应生成化合物6;
[0015]
(6)化合物6与丙二腈发生反应生成化合物7;
[0016]
(7)化合物5与化合物7发生knoevenagel缩合反应生成化合物8;
[0017]
(8)化合物8在强碱的作用下发生水解反应生成目标分子h1;
[0018]
反应式如下:
[0019][0020]
进一步地,步骤(1)包括以下步骤:
[0021]
将化合物1、2-噻吩硼酸和碳酸钾置于双颈烧瓶中,甲苯/乙醇/水作为混合溶剂,
也可使用四氢呋喃/水混合溶剂,抽换氮气3次;氮气氛围下,加入适量的催化剂,升温至80~110℃搅拌反应12~24小时;反应完毕后,冷却至室温,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物通过柱层析分离提纯,洗脱剂比例为石油醚/乙酸乙酯=10:1(v/v),得到化合物2;其中所述的化合物1与2-噻吩硼酸的摩尔比为1:(1~2)。
[0022]
进一步地,步骤(1)所述催化剂为pd(pph3)4、pd(dba)2、pdcl2(dppf)、pd(pph3)2cl2的一种以上。
[0023]
进一步地,步骤(2)包括以下步骤:
[0024]
将化合物2和n,n-二甲基甲酰胺置于双颈烧瓶中,干燥后的1,2-二氯乙烷作为溶剂,抽换氮气3次;氮气氛围下,加入适量的催化剂,随后升温至60~90℃搅拌反应12~48小时;反应完毕后,冷却至室温,加入碳酸钾溶液,再用二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物通过柱层析分离提纯,洗脱剂比例为石油醚/乙酸乙酯/二氯甲烷=25:4:1(v/v),得到化合物3;其中所述的化合物2:n,n-二甲基甲酰胺:催化剂的摩尔比为1:(1~30):(1~15)。
[0025]
进一步地,步骤(2)所述催化剂为三氯氧磷、固体光气、氯化亚砜的一种以上。
[0026]
进一步地,步骤(3)包括以下步骤:
[0027]
将化合物3和四乙基氢氧化铵置于双颈烧瓶中,1,4-二氧六环作为溶剂,抽换氮气3次;氮气氛围下,升温至50~80℃搅拌反应0.5~3小时,随后室温反应1~6小时;反应完毕后,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物通过柱层析分离提纯,洗脱剂比例为石油醚/乙酸乙酯=3:1(v/v),得到化合物4;其中所述的化合物3与四乙基氢氧化铵的摩尔比为1:(1~4)。
[0028]
进一步地,步骤(4)包括以下步骤:
[0029]
将化合物4、溴乙酸乙酯和碳酸钾置于双颈烧瓶中,溶剂为乙腈、甲苯、四氢呋喃、n,n-二甲基甲酰胺的其中一种,抽换氮气3次;氮气氛围下,升温至60~90℃搅拌反应8~24小时;反应完毕后,冷却至室温,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物通过柱层析分离提纯,洗脱剂比例为石油醚/乙酸乙酯=3:1(v/v),得到化合物5;其中所述的化合物4与溴乙酸乙酯的摩尔比为1:(1~5)。
[0030]
进一步地,步骤(5)包括以下步骤:
[0031]
将2-甲基喹啉和溴乙酸乙酯置于双颈烧瓶中,抽换氮气3次;氮气氛围下,升温至100~120℃搅拌反应1.5~4小时;反应完毕后,冷却至室温,甲醇溶解后利用乙酸乙酯作为不良溶剂进行重结晶,所得的固体未经进一步纯化,得到化合物6;其中所述的2-甲基喹啉与溴乙酸乙酯的摩尔比为1:(1~2)。
[0032]
进一步地,步骤(6)包括以下步骤:
[0033]
将化合物6、丙二腈和乙醇钠置于双颈烧瓶中,乙醇作为溶剂,抽换氮气3次;氮气氛围下,室温搅拌反应10~24小时;反应完毕后,浓缩反应液,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物通过柱层析分离提纯,洗脱剂比例为石油醚/乙酸乙酯=1:1(v/v),得到化合物7;其中所述的化合物6与丙二腈的摩尔比为1:(1~4)。
[0034]
进一步地,步骤(7)包括以下步骤:
[0035]
将化合物5、化合物7和哌啶置于双颈烧瓶中,乙腈作为溶剂,抽换氮气3次;氮气氛围下,升温至70~90℃搅拌反应8~24小时;反应完毕后,冷却至室温,加入甲醇至有大量固
体析出,减压抽滤获得粗产物,再用甲醇清洗3遍,得到化合物8;其中所述的化合物5和化合物7的摩尔比为(1~3):1。
[0036]
进一步地,步骤(8)包括以下步骤:
[0037]
将化合物8和强碱置于双颈烧瓶中,此处所述的强碱可使用naoh、koh等强碱,四氢呋喃/水作为溶剂;升温至60~85℃搅拌反应6~24小时;反应完毕后,冷却至室温,浓缩反应液后加入适量的水,再滴加盐酸溶液直至不再有沉淀生成,减压抽滤获得粗产物,粗产物通过柱层析分离提纯,洗脱剂比例为乙酸乙酯/甲醇=2:1(v/v),得到目标分子h1;其中所述的化合物与碱的摩尔比为1:(1~20)。
[0038]
本发明还提供了用于二胺检测的aie柱芳烃荧光探针分子的应用,在溶剂中将探针分子h1与二胺及其它烷基链小分子混合,然后进行荧光光谱检测。
[0039]
进一步地,所述溶剂为乙酸乙酯,所述二胺为烷基二胺。
[0040]
进一步地,所述烷基二胺为1,8-辛二胺、1,10-癸二胺和二胺基十二烷的一种以上。
[0041]
相对于现有技术,本发明具有如下优点和有益效果:
[0042]
(1)本发明首次报道了一种用于二胺检测的aie柱芳烃荧光探针分子,其制备方法简单,具有原料廉价、反应步骤少、条件温和、操作简单和产率高等优点;
[0043]
(2)本发明不仅很好地利用了柱芳烃空腔与客体之间的主-客体作用,还在柱芳烃的两端引入了两个锚固基团。通过多个结合位点以及多种作用力协同作用,该aie柱芳烃荧光探针可以从烷基二胺、单胺、烷基二酸、烷基二醇、二卤代物等众多烷基化合物中选择性检测1,8-辛二胺、1,10-癸二胺和二胺基十二烷,且检测过程操作简便、灵敏度高,可用于环境监测、食品安全检测等。
附图说明
[0044]
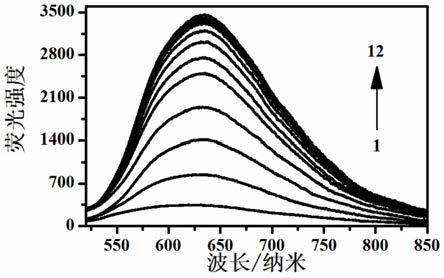
图1为实施例2中h1的荧光强度和1,8-辛二胺加入量变化的关系图;
[0045]
图2为实施例3中h1的荧光强度和1,10-癸二胺加入量变化的关系图;
[0046]
图3为实施例4中h1的荧光强度和二胺基十二烷加入量变化的关系图;
[0047]
图4为实施例5中h1在加入其它客体之后荧光变化谱图。
具体实施方式
[0048]
以下结合实例对本发明的具体实施作进一步说明,但本发明的实施和保护不限于此。需指出的是,以下若有未特别详细说明之过程,均是本领域技术人员可参照现有技术实现或理解的。所用试剂或仪器未注明生产厂商者,视为可以通过市售购买得到的常规产品。
[0049]
实施例1
[0050]
本实施例合成的aie柱芳烃荧光探针的结构式如下所示:
[0051][0052]
具体合成步骤如下:
[0053]
(1)化合物2的合成
[0054][0055]
将化合物1(986mg,1mmol)、2-噻吩硼酸(167mg,1.3mmol)和碳酸钾(483mg,3.5mmol)置于双颈烧瓶中,加入甲苯/乙醇/水(25ml/5ml/2ml)混合溶剂,抽换氮气3次,加入pd(pph3)4(58mg,0.05mmol),升温至90℃搅拌反应24小时。反应完毕后,冷却至室温,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物以石油醚/乙酸乙酯=10:1(v/v)为洗脱剂经硅胶柱层析色谱分离纯化,真空干燥后得到白色固体2(488mg),产率为53%。熔点:183-185℃。
[0056]1hnmr(500mhz,cdcl3)δ7.32-7.30(m,2h),7.22(s,1h),7.07-7.05(m,1h),6.91-6.90(m,1h),6.78(s,1h),6.76(s,1h),6.76(s,1h),6.74(s,1h),6.73(s,1h),6.68(s,1h),6.67(s,1h),6.03(s,1h),3.95(s,2h),3.85(s,2h),3.82(s,2h),3.81(s,2h),3.74(s,2h),3.72(s,3h),3.67(s,3h),3.66(s,3h),3.63(s,3h),3.62(s,3h),3.56(s,3h),3.53(s,3h),3.49(s,3h).
[0057]
(2)化合物3的合成
[0058][0059]
将化合物2(461mg,0.5mmol)和n,n-二甲基甲酰胺(1ml)置于双颈烧瓶中,加入10ml干燥后的1,2-二氯乙烷,抽换氮气3次。氮气氛围下,冰浴条件下加入1ml三氯氧磷作催化剂并搅拌40分钟,随后升温至85℃搅拌反应48小时。反应完毕后,冷却至室温,冰浴条件下加入2mol/l的碳酸钾溶液10ml,再用二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物以石油醚/乙酸乙酯/二氯甲烷=25:4:1(v/v/v)为洗脱剂经硅胶柱层析色谱分离纯化,真空干燥后得到白色固体3(360mg),产率为76%。熔点:144-146℃。
[0060]1hnmr(500mhz,cdcl3)δ9.87(s,1h),7.64(d,j=3.8hz,1h),7.21(s,1h),7.21(s,
1h),6.90(d,j=3.8hz,1h),6.75(s,1h),6.74(s,1h),6.71(s,1h),6.71(s,1h),6.70(s,1h),6.66(s,1h),6.61(s,1h),5.98(s,1h),3.87(s,2h),3.81(s,2h),3.78(s,2h),3.77(s,2h),3.69(s,5h),3.65(s,3h),3.63(s,3h),3.62(s,3h),3.58(s,3h),3.54(s,3h),3.45(s,3h),3.43(s,3h).
[0061]
(3)化合物4的合成
[0062][0063]
称取化合物3(100mg,0.11mmol)和四乙基氢氧化铵(124mg,0.22mmol)置于双颈烧瓶中,加入3ml 1,4-二氧六环,抽换氮气3次。氮气氛围下,升温至60℃搅拌反应半小时,随后室温反应3小时。反应完毕后,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物以石油醚/乙酸乙酯=3:1(v/v)为洗脱剂经硅胶柱层析色谱分离纯化,真空干燥后得到淡黄色固体4(70mg),产率为81%。熔点:119-121℃。
[0064]1hnmr(500mhz,cdcl3)δ9.90(s,1h),7.74(d,j=3.8hz,1h),7.40(s,1h),7.13(s,1h),7.11(d,j=3.8hz,1h),6.99(s,1h),6.81(s,1h),6.79(s,1h),6.76(s,1h),6.68(s,1h),6.66(s,1h),6.57(s,1h),6.44(s,1h),6.43(s,1h),3.88-3.88(m,5h),3.80(s,2h),3.78(s,2h),3.77(s,4h),3.75(s,3h),3.68(s,3h),3.67(s,3h),3.66(s,3h),3.65(s,3h),3.59(s,3h),3.16(s,3h).
[0065]
(4)化合物5的合成
[0066][0067]
将化合物4(600mg,0.74mmol)、溴乙酸乙酯(247mg,1.48mmol)和碳酸钾(413mg,3mmol)置于双颈烧瓶中,加入15ml乙腈,抽换氮气3次。氮气氛围下,升温至80℃搅拌反应24小时。反应完毕后,冷却至室温,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物以石油醚/乙酸乙酯=3:1(v/v)为洗脱剂经硅胶柱层析色谱分离纯化,真空干燥后得到淡黄色固体5(530mg),产率为80%。熔点:185-187℃。
[0068]1hnmr(400mhz,cdcl3)δ9.92(s,1h),7.78(d,j=3.8hz,1h),7.34(s,1h),7.11(d,j=3.8hz,1h),6.92(s,1h),6.87(s,1h),6.86(s,1h),6.82-6.81(m,3h),6.77(s,1h),6.66(s,1h),6.06(s,1h),4.53(s,2h),3.86(s,2h),3.80-3.78(m,10h),3.73-3.70(m,18h),3.62(s,3h),3.50(s,3h),-1.70(t,j=7.0hz,3h).
[0069]
(5)化合物6的合成
[0070][0071]
将2-甲基喹啉(2.86g,20mmol)和溴乙酸乙酯(5g,30mmol)置于双颈烧瓶中,抽换氮气3次。氮气氛围下,升温至110℃搅拌反应3小时。反应完毕后,冷却至室温,甲醇溶解后利用乙酸乙酯作为不良溶剂进行重结晶,所得的固体未经进一步纯化,得到橙黄色固体6(5.2g),产率为84%。
[0072]
(6)化合物7的合成
[0073][0074]
将化合物6(6g,30mmol)、丙二腈(4g,60mmol)和乙醇钠(4g,60mmol)置于双颈烧瓶中,加入35ml乙醇,抽换氮气3次。氮气氛围下,室温搅拌反应16小时。反应完毕后,浓缩反应液,加入二氯甲烷萃取,有机层水洗,干燥,浓缩,粗产物以石油醚/乙酸乙酯=1:1(v/v)为洗脱剂经硅胶柱层析色谱分离纯化,真空干燥后得到黄绿色固体7(1.3g),产率为22%。熔点:151-153℃。1hnmr(400mhz,cdcl3)δ9.03(d,j=8.4hz,1h),7.73-7.69(m,1h),7.44-7.40(m,1h),7.33(d,j=8.8hz,1h),6.80(s,1h),4.94(s,2h),4.30(q,j=7.1hz,2h),2.52(s,3h),1.30(t,j=7.1hz,3h).
[0075]
(7)化合物8的合成
[0076][0077]
将化合物5(300mg,0.26mmol)、化合物7(70mg,0.24mmol)和0.3ml哌啶置于双颈烧瓶中,加入7ml乙腈,抽换氮气3次。氮气氛围下,升温至80℃搅拌反应24小时。反应完毕后,冷却至室温,加入50ml甲醇后有大量固体析出,减压抽滤获得粗产物,再用甲醇(3
×
30ml)清洗粗产物,得到红色固体8(161mg),产率为57%。熔点:255-257℃。1hnmr(400mhz,cdcl3)δ9.14
–
9.12(m,1h),7.76-7.72(m,1h),7.50-7.46(m,2h),7.33-7.32(m,2h),7.30(d,j=3.6hz,1h),7.17(s,1h),6.96(d,j=3.6hz,1h),6.92(s,1h),6.89(s,1h),6.87(s,1h),6.82(s,3h),6.76(s,1h),6.69(d,j=15.4hz,1h),6.62(s,1h),6.17(s,1h),4.96(s,2h),4.51(s,2h),4.36(q,j=7.1hz,2h),3.92(s,2h),3.81-3.78(m,10h),3.73-3.70(m,18h),3.61(s,3h),3.55(s,3h),1.34(t,j=7.1hz,3h),-1.67(t,j=6.7hz,3h).
[0078]
(8)h1的合成
[0079][0080]
将化合物8(190mg,0.16mmol)和氢氧化钠(66mg,1.64mmol)置于双颈烧瓶中,加入四氢呋喃/水(25ml/5ml)混合溶剂,升温至73℃搅拌反应12小时。反应完毕后,冷却至室温,浓缩反应液后加入25ml水,再滴加盐酸溶液至不再有沉淀生成,减压抽滤获得粗产物,粗产物以乙酸乙酯/甲醇=2:1(v/v)为洗脱剂经硅胶柱层析色谱分离纯化,真空干燥后得到红色固体h1(148mg),产率为82%。熔点:176-178℃。
[0081]1hnmr(500mhz,cdcl3)δ9.07(d,j=8.3hz,1h),7.75-7.72(m,1h),7.52-7.39(m,4h),7.27(s,1h),7.15(s,1h),6.99-6.99(m,1h),6.84(s,1h),6.78(s,1h),6.76(s,1h),6.69-6.59(m,5h),6.55(s,1h),6.15(s,1h),4.95(s,2h),4.43(s,2h),3.93(s,2h),3.84(s,2h),3.77(s,2h),3.76(s,2h),3.74-3.73(m,5h),3.68(s,3h),3.67(s,3h),3.63(s,3h),3.58(s,3h),3.48-3.45(m,9h).
[0082]
实施例2
[0083]
将h1溶解于乙酸乙酯中,获得探针母液(1
×
10-3
mol/l),随后取50微升该母液并利用乙酸乙酯将其稀释至2ml,得到2.5
×
10-5
mol/l的h1溶液。照此方法配置12个样品,然后在这些样品中分别加入0微升(1)、2微升(2)、4微升(3)、6微升(4)、8微升(5)、10微升(6)、12微升(7)、14微升(8)、16微升(9)、18微升(10)、20微升(11)、24微升(12)的5
×
10-3
mol/l的1,8-辛二胺溶液,混合均匀后静置5分钟,分别测定这些样品的荧光强度,见图1。测定结果表明:随着1,8-辛二胺加入量的不断增加,探针分子h1的荧光发射强度也在不断增加。通过线性拟合的结果可以计算得知h1对1,8-辛二胺的检测限为16.8
±
0.1nm。
[0084]
实施例3
[0085]
将h1溶解于乙酸乙酯中,获得探针母液(1
×
10-3
mol/l),随后取50微升该母液并利用乙酸乙酯将其稀释至2ml,得到2.5
×
10-5
mol/l的h1溶液。照此方法配置13个样品,然后在这些样品中分别加入0微升(a)、1微升(b)、2微升(c)、4微升(d)、6微升(e)、8微升(f)、10微升(g)、12微升(h)、14微升(i)、16微升(j)、18微升(k)、20微升(l)、26微升(m)的5
×
10-3
mol/l的1,10-癸二胺溶液,混合均匀后静置5分钟,分别测定这些样品的荧光强度,见图2。测定结果表明:随着1,10-癸二胺加入量的不断增加,探针分子h1的荧光发射强度也在不断增加。通过线性拟合的结果可以计算得知h1对1,10-癸二胺的检测限为9.9
±
0.1nm。
[0086]
实施例4
[0087]
将h1溶解于乙酸乙酯中,获得探针母液(1
×
10-3
mol/l),随后取50微升该母液并利用乙酸乙酯将其稀释至2ml,得到2.5
×
10-5
mol/l的h1溶液。照此方法配置14个样品,然后在这些样品中分别加入0微升(a)、1微升(b)、2微升(c)、3微升(d)、4微升(e)、6微升(f)、8微升(g)、10微升(h)、12微升(i)、14微升(j)、16微升(k)、18微升(l)、20微升(m)、26微升(n)的5
×
10-3
mol/l的二胺基十二烷溶液,混合均匀后静置5分钟,分别测定这些样品的荧光强度,
见图3。测定结果表明:随着二胺基十二烷加入量的不断增加,探针分子h1的荧光发射强度也在不断增加。通过线性拟合的结果可以计算得知h1对二胺基十二烷的检测限为7.6
±
0.2nm。
[0088]
实施例5
[0089]
将h1溶解于乙酸乙酯中,获得探针母液(1
×
10-3
mol/l),随后取50微升该母液并利用乙酸乙酯将其稀释至2ml,得到2.5
×
10-5
mol/l的h1溶液。照此方法配置9个样品,以其中一个样品作空白对照,然后在其余8个样品中分别加入20微升的浓度均为5
×
10-3
mol/l的正辛胺、乙二胺、1,4-丁二胺、1,6-己二胺、辛二腈、1,8-辛二醇、1,8-辛二酸和1,8-二溴辛烷溶液,混合均匀后静置5分钟,分别测定这些样品的荧光强度,然后与实施例2、实施例3、实施例4分别得到的相应的荧光发射曲线对比,如图4所示。测定结果表明:加入的这些客体分子都不能使探针分子h1的荧光发射强度发生很明显的变化,从而证明只有客体在同时符合拥有两个胺基和烷基链长度合适的条件下,客体分子才能与探针分子h1在进行主客体结合后利用多个结合位点以及多种作用力协同作用实现h1荧光的增强,进而完成对1,8-辛二胺、1,10-癸二胺和二胺基十二烷的选择性识别。
[0090]
以上实施例仅为本发明较优的实施方式,仅用于解释本发明,而非限制本发明,本领域技术人员在未脱离本发明精神实质下所作的改变、替换、修饰等均应属于本发明的保护范围。