1.本技术涉及生物材料领域,具体而言,涉及一种二聚核苷酸的合成方法。
背景技术:
2.寡核苷酸通常是指一类碱基含量较少的短链核苷酸的总称。
3.由于寡核苷酸容易与它们的互补对连接,所以寡核苷酸常作为探针确定dna或rna的结构,并且经常用于基因芯片、电泳、荧光原位杂交等过程中。
4.目前,被广泛使用的寡聚核苷酸的制备方法是固相亚磷酰胺法。其原理在于核苷3
′
固定在固相的载体上(例如,可控微孔玻璃珠/cpg),并在5
′
上连接一个保护基团(例如,二甲氧基三苯甲基/dmt),使合成的方向由带合成引物的3
′
端向5
′
端合成。并且,在核苷酸聚合的结构中,相邻的核苷酸通过3
′→5′
磷酸二酯键连接。
5.该方案具有高效、快速的偶联以及起始反应物比较稳定的特点。但是,该方法也存在受设备的局限而限制规模化的生产。另外,近来常通过合成仪按设计自动连接氨基酸(多肽合成仪)或核苷酸(dna、rna合成仪)合成期望结构,但是其产量不高。
技术实现要素:
6.本技术提供了一种二聚核苷酸及其合成方法。该二聚核苷酸可以可通过合成简便的方法进行合成,并且提供了一种在常见的以3
′‑5′
磷酸二酯键连接的寡聚核苷酸之外的低聚核苷酸生物材料。其能够被用于丰富化合物库,从而有助于在药物的高通量筛选方面的应用。
7.本技术是这样实现的:
8.在第一方面,本技术示例提出了一种二聚核苷酸的合成方法,其包括:使核苷的2
′
羟基和脱氧核苷的5
′
羟基通过一个2
′‑5′
磷酸二酯键缩聚而成二聚核苷酸,二聚核苷酸具有下式1-1结构:
[0009][0010]
其中r1、r2分别独立地选自天然碱基、经过修饰的碱基、天然碱基的保护形式或经过修饰的碱基的保护形式,碱基包括腺嘌呤(a)、鸟嘌呤(g)、胞嘧啶(c)、胸腺嘧啶(t)或尿嘧啶(u);
[0011]
a1和a2不同且分别选择自羟基或氢。
[0012]
根据本技术的一些示例,天然碱基的保护形式包括n-6-苯甲酰基腺嘌呤(abz)、n-2-异丁酰基鸟嘌呤(gibu)或n-4-乙酰基胞嘧啶(cac)。
[0013]
根据本技术的一些示例,a1为羟基,a2为氢,且脱氧核苷的由a1表示的3
′
羟基是保
护形式,且5
′
羟基是保护形式。例如5
′
羟基经过用于提供形成磷酸二酯键中的磷元素的亚磷酰胺化基团的保护。
[0014]
根据本技术的一些示例,脱氧核苷具有如下式i的结构;
[0015]
或者,或者,或者,或者,
[0016]
其中x表示用于保护羟基的基团,且4,4
′‑
二甲氧基三苯甲基(dmtr)、或4-甲氧基三苯甲基(mmtr)或三苯甲基(tr)。
[0017]
根据本技术的一些示例,核苷的3
′
羟基、5
′
羟基与硅醚结合而以保护形式存在,硅醚包括三甲基硅醚、叔丁基二甲基硅醚或三异丙基硅醚。
[0018]
根据本技术的一些示例,核苷的3
′
羟基、5
′
羟基分别通过三异丙基硅醚进行保护,核苷具有如下式ii的结构:
[0019][0020]
根据本技术的一些示例,a1为羟基,a2为氢,合成方法按照下述反应方程式进行:
[0021][0022]
根据本技术的一些示例,在上述反应方程式中,脱保护是分步完成的且包括先脱除dmtr基团,再脱除三异丙基硅醚基团;
[0023]
其中,dmtr基团的脱除是在dca(二氯乙酸)和dcm的溶液体系中发生的,三异丙基硅醚基团的脱除是在py(吡咯)、hf和dcm的溶液体系中发生的;
[0024]
可选地,合成方法包括:在按照上述反应方程式进行反应之后,对料液进行纯化操
作。
[0025]
根据本技术的一些示例,合成方法按照下述反应方程式进行:
[0026][0027]
在第二方面,本技术示例提出了一种二聚核苷酸,二聚核苷酸由核苷的2
′
羟基和脱氧核苷的5
′
羟基通过一个2
′‑5′
磷酸二酯键缩聚而成,二聚核苷酸具有下式1-1结构:
[0028]
其中r1、r2分别独立地选自天然碱基、经过修饰的碱基、天然碱基的保护形式或经过修饰的碱基的保护形式,碱基包括腺嘌呤(a)、鸟嘌呤(g)、胞嘧啶(c)、胸腺嘧啶(t)或尿嘧啶(u);
[0029]
a1和a2不同且分别选择自羟基或氢。
[0030]
本技术实施例提供的二聚核苷酸作为一种单链脱氧寡核苷酸在制备狂犬疫苗、乙型肝炎疫苗、流感病毒疫苗和其它蛋白类疫苗的生物制剂等方面具有潜在的用途。
附图说明
[0031]
为了更清楚地说明本技术实施例或现有技术中的技术方案,以下将对实施例或现有技术描述中所需要使用的附图作简单地介绍。
[0032]
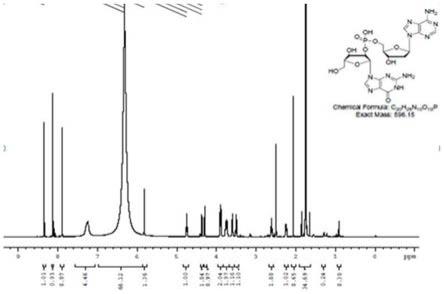
图1为实施例1中制备的二聚核苷酸的1h-nmp谱图;
[0033]
图2为实施例2中制备的二聚核苷酸的1h-nmp谱图。
具体实施方式
[0034]
传统药物通常是直接作用于致病蛋白,而反义药物则作用于产生致病蛋白的基因。反义药物通过抑制前述基因的表达(基因沉默)而阻止致病蛋白的产生,从而达到预防或治疗的效果。
[0035]
与传统药物比较,反义药物更具有选择性,因此也更高效、低毒。迄今为止,反义药物已经被广泛应用于多种疾病—如传染病、炎症、心血管疾病及肿瘤等—的治疗。
[0036]
寡聚核苷酸在反义药物的研究中具有重要的位置。因此,反义药物一般指的是反义寡聚核苷酸(antisense oligonucleotide,简称为ason)药物。反义寡聚核苷酸可以通过
序列特异地与靶基因dna或mrna结合,从而抑制该基因表达。其是一种能够在基因水平调控的分子药物。
[0037]
作为一种寡聚核苷酸中的基础性分子,目前,二聚核苷酸通常来源于寡核苷酸类化合物的合成工艺中的最起始的产物。并且,在这些二聚核苷酸通常的连接方式为3'-5'的顺序连接。
[0038]
为了方便理解和描述连接方式,碳按照下述方式编号。
[0039]
核苷中的五碳糖上的碳编号如下式所示。
[0040][0041]2′‑
脱氧核苷的五碳糖的碳编号如下式所示。
[0042][0043]
常见的3'-5'连接的二聚核苷酸的获得方式是采用合成仪按设计自动连接核苷酸(dna、rna合成仪),从而合成期望结构。但是其产量不高且容易受到设备的限制而不易进行工业化的大规模生产。
[0044]
有鉴于此,在本技术,发明人提出了一种新的二聚核苷酸,其具有区别于3'-5'的新的连接方式—2'-5'连接。并且,该二聚核苷酸的2'-5'连接形成磷酸二酯键。示例中,二聚核苷酸通过由核苷的2
′
羟基和脱氧核苷的5
′
羟基通过一个2
′‑5′
磷酸二酯键缩聚而成。
[0045]
下面将结合实施例对本技术的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本技术,而不应视为限制本技术的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0046]
以下针对本技术实施例的二聚核苷酸及其合成方法进行具体说明。
[0047]
本技术示例中的二聚核苷酸是一种寡聚核苷酸,或者也可以被称之为低聚核苷酸。该二聚核苷酸是由两个单体通过磷酸二酯键结合/缩聚而成的二聚体。并且,如前文所述,该两个单体中的一个为脱氧核苷,另一个核苷。
[0048]
示例性地,核苷的结构为
[0049]
示例性地,脱氧核苷的结构为或者
[0050]
其中,r1和r2表示的是核碱基(nucleobase)或者也简称为碱基(base)或其衍生物。例如,r1和r2可以表示腺嘌呤(adenine,缩写为a)、鸟嘌呤(guanine,缩写为g)、胞嘧啶(cytosine,缩写为c)、胸腺嘧啶(thymine,缩写为t)或尿嘧啶(uracil,缩写为u)。即这些碱基可以是天然碱基。
[0051]
或者,r1核r1表示的是碱基衍生物,例如可以是天然碱基的保护形式、或者经过修饰的碱基、或者经过修饰的碱基的保护形式。
[0052]
其中的保护形式主要是指碱基的活性基团被通过引入其他惰性基团/保护基团进行保护且能够在适当的条件下脱除该惰性基团,从而恢复原活性基团。其中,碱基的活性基团例如是胺基;示例性地,碱基的活性基团例如是腺嘌呤的伯胺基、鸟嘌呤的伯胺基、胞嘧啶的伯胺基。
[0053]
作为示例,腺嘌呤的伯胺基的保护形式可以是n-6-苯甲酰基腺嘌呤(abz,cas登录号为4005-49-6);其中的保护基团是苯甲酰基。鸟嘌呤的伯氨基的保护形式例如是n-2-异丁酰鸟嘌呤(gibu,cas登录号为21047-89-2);其中的保护基团为异丁酰基。胞嘧啶的伯胺基的保护形式可以是n-4-乙酰基胞嘧啶(cac,cas登录号为26661-13-2);其中的保护基团是乙酰基。
[0054]
因此,该二聚核苷酸的结构可以表示为如下式1-1所示。
[0055][0056]
从结构式中的连接方式而言,其可以被描述为一种r[2
′
,5
′
]p(3
′
or2
′
)dr分子。其中r1、r2分别独立地选自天然碱基、经过修饰的碱基、天然碱基的保护形式或经过修饰的碱基的保护形式。并且,a1和a2是不同且位于脱氧核苷的核糖的碳环的2
′
、3
′
上的氢或羟基。即,当a1表示羟基(oh)时,a2表示氢;当a1表示羟基氢时,a2表示羟基(oh)。换言之,本技术示例中的二聚核苷酸中的脱氧核苷的脱氧位置是2
′
或者3
′
碳上。
[0057]
示例性地,二聚核苷酸可以由下述的两结构所表示:
[0058][0059]
为了本领域技术人员更易于获得上述二聚核苷酸,示例中给出了一种二聚核苷酸的非固相合成的制备方案。作为对比,目前使用得较多的方式是固相合成,并且该方案需要使用固相载体—cpg(controlled pore glass),即微孔的二氧化硅材质的玻璃球。
[0060]
本技术示例中,将对脱氧核苷和核苷进行基团修饰,从而形成两中间体,并以该两中间体为起始物进行反应,制备二聚核苷酸。该合成方法可以按照下述的反应方程式进行:
[0061]
[0062][0063]
脱氧核苷中间体i
[0064]2′‑
脱氧核苷先进行碱基保护和3
′
羟基保护,然后将磷引入结构中,从而获得中间体z-1;其结构如下式所示。
[0065]
其中的r1可以是abz、gibu、cac或t。
[0066]
其中的3
′
羟基保护可以通过在脱氧核苷中引入4,4
′‑
o-二甲氧基三苯甲基(dmtr;4,4
′‑
o-二甲氧基三苯甲基氯,cas:40615-36-9)进行保护,后续则可以通过使用四丁基氯
化铵(tbaf)或dca(二氯乙酸)进行脱保护。
[0067]
磷的引入可通过使用亚磷酰化试剂或称磷试剂实现。该磷试剂例如是双(二异丙基氨基)(2-氰基乙氧基)膦(cas:102691-36-1)进行反应。例如可以将经过3
′
羟基保护的脱氧核苷在乙腈体系中,加入四氮唑进行反应。该磷试剂也可以采用2-氰乙基-n,n-二异丙基氯代亚磷酰胺(cas:89992-70-1)代替,其反应活性高,容易与醇羟基反应进行缩合。
[0068]
核苷中间体ii
[0069]
其中,核苷部分先进行碱基保护和3
′
,5
′
羟基保护,从而获得中间体z-2;其结构如下式所示。
[0070]
该式中的r2可以式abz、gibu、cac或u。
[0071]
其中,3
′
,5
′
羟基的保护是通过在核苷的五元杂环的主体结构中引入tips(1,1,3,3-四异丙基二硅氧烷,cas:18043-71-5)达到在合成中对羟基保护。例如,将核苷溶解在吡啶中,然后加入tips室温搅拌反应获得式z-2所示的结构。然后冷却至0℃、加水搅拌,再用乙酸乙酯进行分液,再将水层用乙酸乙酯萃取;进一步可以通过柱层析的方式纯化。后续在适当的步骤中,tips可以被脱去,例如通过维蒂希反应(witting reaction)脱除tips。
[0072]
在获得上述两个中间体后,将中间体z-1和中间体z-2缩聚反应,然后经过氧化、脱保护,再水解产生二聚核苷酸。
[0073]
示例性地,中间体z-1和中间体z-2在二氯甲烷(dcm)体系中,以四氮唑为催化剂进行聚合反应从而获得下式iii的结构:
[0074]
反应过程中可以控制温度在25℃至30℃。
[0075]
四氮唑是一种弱酸性试剂且用以催化使磷试剂活化,从而达到中间体i中的磷试剂与中间体ii的2羟基反应。当磷试剂为2-氰乙基-n,n-二异丙基氯代亚磷酰胺时,也可以不使用上述的弱酸性试剂—四氮唑。另外,四氮唑还可以由4,5-二氰基咪唑以及吡啶三氟乙酸盐等替代;或者,还可以使用二氮唑和二氯甲烷。
[0076]
式iii结构继续在dcm体系中,以过氧化氢叔丁基作为氧化剂对磷进行氧化,从而获得式iv结构的分子:
[0077][0078]
随后脱除3
′
的羟基保护基团(如上述的dmtr)和3
′‑5′
的羟基保护基团(如上述的tips);然后在氨水中水解将碱基r1和r2中如果存在的胺保护基团去除,同时将氰乙氧基去除。
[0079]
进一步地还可以通过柱层析分离的方式进行精制。一些示例中,选择离子交换树脂进行柱层析,进一步地还可以继续选择使用c18色谱柱进行梯度洗脱,从而获得纯度更高的前述二聚核苷酸。
[0080]
以下结合实施例对本技术的二聚核苷酸及其合成方法作进一步的详细描述。
[0081]
实施例1
[0082]
原料:
[0083]
化合物1
[0084]
化合物2
[0085]
原料处理:45.23g(0.9eq)的化合物1,和34.9g化合物2,用350ml的二氯甲烷dcm带水进行除水干燥。
[0086]
第一步骤、在烧瓶内加入上述化合物1和化合物2,并将四氮唑4.1g(1.0eq)加入其中。在26℃反应14h。纯化:用碳酸氢钠和氯化钠水溶液萃取,有机相进行干燥、过滤、浓缩;再用17倍质量填充的硅胶层析进行洗脱,其中的洗脱液是10倍柱体积的hep/ea=1/2。经过洗脱收集含化合物3(含量88.09%)的洗脱液,再将洗脱液浓缩干得40.0g固体,化合物3的纯度:97.30%。
[0087]
化合物3的结构
[0088]
第二步骤、将34.5g(1.0ep)的化合物3,与345ml二氯甲烷dcm、7.7ml(1.5eq)的过氧化叔丁醇在容器中混合,并在25
±
3℃反应通过tlc监控直至无化合物3为反应终点。纯化:2.5eq硫代硫酸钠水溶液淬灭,水洗2次,得纯度为97.22%的化合物4。
[0089]
化合物4的结构
[0090]
第三步骤、将25g化学物4用250mldcm溶解,并加入5.85g(2.5eq)的二氯乙酸dca、3.29g(10.0eq)的水。然后在20
±
5℃的温度下反应4h。纯化:反应液加碳酸氢钠冰水溶液洗2次,获得化合物5的粗品(直接投下一步反应)。
[0091]
化合物5的结构
[0092]
第四步骤、将19g的化合物5粗品和9.32g的tbaf用100ml的四氢呋喃thf溶解,在25
±
2℃反应1h,获得化合物6(转化率71.88%),反应结束后反应液直接投下一步。
[0093]
化合物6的结构
[0094]
第五步骤、向第四步骤中的反应液中加入200ml氨水,升温到35
±
2℃水解反应12h
后,获得化合物7:45.27%。
[0095]
反应液加200ml水稀释,于25-30℃浓缩至无冷凝液滴下,水相用ea萃取2次,浓缩后化合物7纯度:43.17%。
[0096]
化合物7的结构
[0097]
纯化:c18色谱柱,以甲醇和10mm醋酸铵为流动相进行梯度洗脱。下柱液40℃减压浓缩带干刮粉得到产品5g。通过hplc分析纯度为99.83%,1h-nmp测试结构符合(如图1所示)、31p-nmp99.80%。
[0098]
实施例2
[0099]
原料处理:化合物1加入31.0g,化合物2加入39.41g(0.9eq)用dcm带干。
[0100]
第一步骤,将化合物1和2,用500ml的dcm溶解,加入四氮唑3.85g(1.0eq),25
±
2℃反应12h后。然后水溶液萃取,有机相干燥过滤浓缩,16倍装填硅胶层析,8倍柱体积hep/ea=1/1.5洗脱收集下柱液,减压浓缩干得化合物3产品57.0g,纯度:83.97%;化合物2的转化率85.29%。
[0101][0102]
第二步骤,48.45g(1.0eq)的化合物3用dcm 485ml溶解,并加入过氧化叔丁醇11.6ml(1.5eq),于25
±
2℃反应1h后tlc监控无原料,使用2.5eq硫代硫酸钠水溶液淬灭,水洗2次,得产品60.0g化合物4(纯度:84.59%)粗品直接投料使用。60.0g的化合物4粗品,加入300mlthf溶解,加入tbaf24.62g(2.0eq),于25
±
2℃反应3.5h得到纯度为73.26%的化合物5。反应结束后反应液直接投下一步。
[0103][0104][0105]
第三步骤,向含化合物5的反应液中直接加入600ml氨水,升温到35
±
2℃反应14h后,获得化合物6:42.59%。
[0106][0107]
纯化:反应液加400ml水稀释,于25-30℃浓缩至无冷凝液滴下,水相用ea萃取2次,浓缩后分析,纯度:45.32%。
[0108]
进一步通过c18色谱进行纯化:
[0109]
柱dac250*50mm,流速:100ml/min;流动相:a:10mm醋酸铵b:甲醇;梯度洗脱制度如
下表所示。
[0110][0111]
将洗脱的下柱液在35℃下减压浓缩得到固体。hplc检测化合物6的纯度为99.3%,1h-nmp如图2所示。
[0112]
以上所述仅为本技术的优选实施例而已,并不用于限制本技术,对于本领域的技术人员来说,本技术可以有各种更改和变化。凡在本技术的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本技术的保护范围之内。