利用有机模板合成菱沸石
1.本技术要求2019年6月13日提交的美国临时申请号62/860,908的优先权权益,所述专利以引用方式整体并入本文。
技术领域
2.本发明大体涉及使用一种或多种有机结构导向剂(osda)生产的具有cha结构的初合成微孔材料,所得菱沸石(cha),和将菱沸石用于选择性催化还原(scr)的方法。
3.发明背景
4.早就已知氮氧化物(nox)是污染气体,主要是由于它们的腐蚀性作用。实际上,它们是酸雨成因的主要原因。nox污染的主要原因是它们在柴油汽车以及诸如燃煤发电厂和汽轮机的固定污染源的排气中的排放。为了避免这些有害排放,scr被采用并且涉及在nox转化成氮气和水中使用沸石催化剂。
5.因此,持续需要改进的微孔晶体材料,其具有提高的生产经济性、性能和水热稳定性以允许选择性催化还原排气中的nox。
6.铝硅酸盐cha型沸石是汽车应用中no
x
消除的商用选择性催化还原(scr)系统中的重要组分。铝硅酸盐cha型沸石可在缺乏或存在osda的情况下生产。一般而言,不存在osda时生产的菱沸石的组成范围、诸如二氧化硅与氧化铝摩尔比可能受限。
7.可在存在一种或多种osda、有时被称为一种或多种模板和共模板时生产菱沸石。一种或多种osda的使用一般允许更宽范围的菱沸石组成、诸如二氧化硅与氧化铝比率。然而,用作菱沸石模板的某些osda已知对于大规模商用而言成本较高。
8.因此,需要可代替传统模板化材料使用或除传统模板化材料外又使用的新osda,具体而言用于制造菱沸石。还需要用较便宜的有机模板可靠地替换昂贵的有机模板。更具体而言,需要改进的osda,其允许提高的生产经济性、性能和水热稳定性,并且最终允许选择性催化还原排气中的nox。
技术实现要素:
9.为了解决前述需要,公开了初合成的微孔材料,其具有cha结构并且包含具有如下季铵阳离子的通用结构的osda:
[0010][0011]
其中r1为衍生或非衍生的c1-c5烷基链,并且r2为衍生或非衍生的c2-c5烷基链,
[0012]
其中x为h(氢)或者一个或多个衍生或非衍生的c1-c3烷基取代基,所述c1-c3烷基取代基连接至构成六氢-1h-氮杂卓鎓环的碳原子的任何组合。
[0013]
还公开了一种通过煅烧本文所述的初合成微孔材料制成的微孔晶体材料。
[0014]
还公开了一种选择性催化还原排气中的氮氧化物的方法。在实施方案中,所述方法包括使排气与包含本文所述的微孔晶体材料的制品至少部分接触。接触步骤可在氨、脲、生成氨的化合物或烃化合物的存在下进行。
[0015]
在实施方案中,公开了一种制造微孔晶体材料的方法,该微孔晶体材料具有至少10、诸如10至50的二氧化硅与氧化铝摩尔比(sar),并且使用具有如下季铵阳离子的通用结构的第一osda来制造:
[0016][0017]
其中r1为衍生或非衍生的c1-c5烷基链,并且r2为衍生或非衍生的c2-c5烷基链,
[0018]
其中x为h(氢)或者一个或多个衍生或非衍生的c1-c3烷基取代基,所述c1-c3烷基取代基连接至构成六氢-1h-氮杂卓鎓环的碳原子的任何组合。
[0019]
在实施方案中,所述方法包括混合氧化铝、二氧化硅、碱金属、第一osda、任选的第二osda和水的来源以形成凝胶,在高压釜中加热凝胶以形成晶体cha产物,并且煅烧所述cha产物。
[0020]
附图简述
[0021]
附图并入本说明书并构成本说明书的一部分。
[0022]
图1是根据实施例1制成的发明性菱沸石产物的x射线衍射图案。
[0023]
图2是根据实施例2制成的发明性菱沸石产物的x射线衍射图案。
[0024]
图3是根据实施例4制成的发明性菱沸石产物的x射线衍射图案。
[0025]
图4是根据实施例6制成的发明性菱沸石产物的x射线衍射图案。
[0026]
图5是根据实施例8制成的发明性菱沸石产物的x射线衍射图案。
[0027]
图6是根据比较实施例1制成的菱沸石产物的x射线衍射图案。
[0028]
图7是根据比较实施例2制成的菱沸石产物的x射线衍射图案。
[0029]
图8是根据比较实施例3制成的菱沸石产物的x射线衍射图案。
[0030]
图9是根据比较实施例4制成的菱沸石产物的x射线衍射图案。
[0031]
发明详述
[0032]
定义
[0033]“初合成”意指在煅烧前、是结晶凝胶的固体产物的微孔晶体材料。
[0034]“水热稳定”意指在暴露于升高的温度和/或湿度条件(相比于室温)一定时间段之后,具有保持初始表面积和/或微孔体积的一定百分比的能力。例如,在一个实施方案中,意指在暴露于模拟汽车排气中存在的条件的条件,诸如在至多10体积百分比(体积%)水蒸气的存在下,至多900℃的温度、包括700至900℃的温度,持续至多1小时、或甚至至多16小时、诸如1至16小时的时间之后,保持至少65%、诸如至少70%、至少80%、至少90%、或甚至至少95%的其表面积、微孔体积和xrd图案强度。
[0035]“初始表面积”意指新鲜制成的晶体材料在暴露于任何老化条件之前的表面积。
[0036]“微孔体积”用来指示具有小于20埃的直径的孔的总体积。“初始微孔体积”意指新
鲜制成的晶体材料在暴露于任何老化条件之前的微孔体积。微孔体积的评价尤其来源于bet测量技术,利用文献(journal of catalysis 3,32(1964))中所描述的称为t图法(或有时仅被称为t法)的评估方法。
[0037]
本文中“中孔体积”是具有大于20埃至600埃的限值的直径的孔的体积。
[0038]
类似地,“微孔面积”指的是小于20埃的孔的表面积,并且“中孔面积”指的是介于20埃与600埃之间的孔的表面积。
[0039]“由国际沸石协会结构委员会定义”意指包括但不限于以全文引用方式并入本文的“atlas of zeolite framework types”,baerlocher等人编,修订第六版(elsevier 2007)中所描述的结构的那些结构。
[0040]“双6环(d6r)”是以全文引用方式并入本文的“atlas of zeolite framework types”,baerlocher等人编,修订第六版(elsevier 2007)中所描述的一种结构性构造单元。
[0041]“选择性催化还原”或“scr”指的是在氧气的存在下将no
x
(通常利用脲和/或氨)还原形成氮气和h2o。
[0042]“排气”指的是工业过程或运行中和由内燃机所形成的、诸如来自任何形式的机动车辆的任何废气。
[0043]
如本文中所使用的用语“选自”指的是对单个组分或两个(或更多个)组分的组合进行选择。例如,本文所述的催化活性金属可选自铜和铁,这意指金属可包含铜或铁,或铜和铁的组合。
[0044]
在第一实施方案中,描述了一种初合成微孔材料,其具有cha结构并且包含至少一种具有如下季铵阳离子的通用结构的osda:
[0045][0046]
其中r1为衍生或非衍生的c1-c5烷基链,且
[0047]
r2为衍生或非衍生的c2-c5烷基链,并且
[0048]
其中x为h(氢)或者一个或多个衍生或非衍生的c1-c3烷基取代基,所述c1-c3烷基取代基连接至构成六氢-1h-氮杂卓鎓环的碳原子的任何组合。
[0049]
在本文所述的初合成微孔材料的实施方案中,r1和r2中的至少一者为乙基。
[0050]
在实施方案中,至少一种osda是氢氧化物或者选自氟化物、氯化物、溴化物、碘化物或它们的混合物的盐。
[0051]
如指示的,初合成形式的微孔晶体材料含有一种或多种用于生产微孔晶体材料的osda。在煅烧前,一种或多种、诸如两种osda在初合成微孔晶体材料中的存在可借助液相色谱来测定。将已知重量的初合成沸石样品溶解于氢氟酸中以将一种或多种osda提取至溶液中。用液相色谱分析所述溶液以测定一种或多种osda的浓度。然后由溶液中一种或多种osda的浓度和初合成沸石样品的重量计算初合成沸石中一种或多种osda的重量百分比。本文所述的初合成微孔材料包含至少0.01重量%、诸如0.01至30重量%、诸如0.01至25重
量%、0.1重量%至22%、或1.0至20重量%的量的季铵阳离子材料。在这些范围中的任何版本也是可能的,诸如0.01至22重量%、0.1至20重量%或0.01至1.0重量%。
[0052]
申请人已意外地发现,使用osda(模板)可导致形成cha型沸石,其中osda具有如下的通用季铵阳离子结构:
[0053][0054]
其中r1为衍生或非衍生的c1-c5烷基链,诸如乙基;且r2为衍生或非衍生的c2-c5烷基链,诸如乙基;并且其中x为h(氢)或者一个或多个衍生或非衍生的c1-c3烷基取代基,所述c1-c3烷基取代基连接至构成六氢-1h-氮杂卓鎓环的碳原子的任何组合。
[0055]
第一osda可以氢氧化物形式或盐形式使用,盐形式包括但不限于氟化物、氯化物、溴化物、碘化物或乙酸盐形式,或它们的混合物。
[0056]
申请人已意外地发现,使用第一osda和以少于通常实践量使用的第二osda可导致形成cha型沸石。第一osda具有如下的通用季铵阳离子结构:
[0057][0058]
其中r1为衍生或非衍生的c1-c5烷基链,诸如乙基;且r2为衍生或非衍生的c2-c5烷基链,也诸如乙基;并且其中x为h(氢)或者一个或多个衍生或非衍生的c1-c3烷基取代基,所述c1-c3烷基取代基连接至构成六氢-1h-氮杂卓鎓环的碳原子的任何组合。
[0059]
在实施方案中,第一osda可以氢氧化物形式或盐形式使用,盐形式包括但不限于氟化物、氯化物、溴化物、碘化物或乙酸盐形式,或它们的混合物。
[0060]
在实施方案中,第二osda是呈氢氧化物形式或盐形式的n,n,n-三甲基-1-金刚烷基铵、n-乙基-n,n-二甲基环己基铵或苄基三甲基铵,盐形式包括但不限于氟化物、氯化物、溴化物、碘化物或乙酸盐形式,或它们的混合物。
[0061]
公开了一种使用一种或多种osda生产的,具有至少10、诸如10至50的二氧化硅与氧化铝摩尔比(sar)的可用微孔晶体材料。所公开的材料特别可用于选择性催化还原氮氧化物。
[0062]
在实施方案中,微孔晶体材料可包含具有cha(菱沸石)的结构代码的晶体结构。具有cha骨架类型的沸石类材料是含有双六环和笼的三维8元环孔/通道体系。
[0063]
在实施方案中,本文所述的初合成微孔材料可被用来制造微孔晶体材料,通过煅烧初合成微孔材料来制成微孔晶体材料。
[0064]
在实施方案中,微孔晶体材料还可包含至少一种催化活性金属,诸如铜或铁。在实施方案中,催化活性金属包含以至少1重量%、诸如1-10重量%的cuo存在的铜cu。在实施方
案中,催化活性金属包含以至少0.2重量%、诸如0.2-10重量%的fe2o3存在的铁fe。
[0065]
还公开了一种选择性催化还原排气中的氮氧化物的方法。在实施方案中,所述方法包括使排气与包含本文所述的微孔晶体材料的制品至少部分接触。接触步骤通常在氨、脲、生成氨的化合物或烃化合物的存在下进行。
[0066]
还描述了一种制造本文所述的微孔晶体材料的方法。在实施方案中,所述方法包括混合氧化铝、二氧化硅、含碱金属的添加剂、一种或多种有机结构导向剂和水的来源以形成凝胶。所述方法进一步包括在高压釜中加热凝胶以形成晶体cha产物,并且煅烧所述cha产物。
[0067]
在实施方案中,所述方法进一步包括通过液相或固相离子交换、浸渍、直接合成或它们的组合,将至少一种催化活性金属、诸如铜或铁引入微孔晶体材料中。
[0068]
在实施方案中,催化活性金属包含以至少1重量%、诸如1-10重量%的cuo存在的铜cu。在实施方案中,催化活性金属包含以至少0.2重量%、诸如0.2-10重量%的fe2o3存在的铁fe。
[0069]
本文所述的方法使用一种或多种osda以形成所得沸石材料。第一osda具有如下季铵阳离子的通用结构:
[0070][0071]
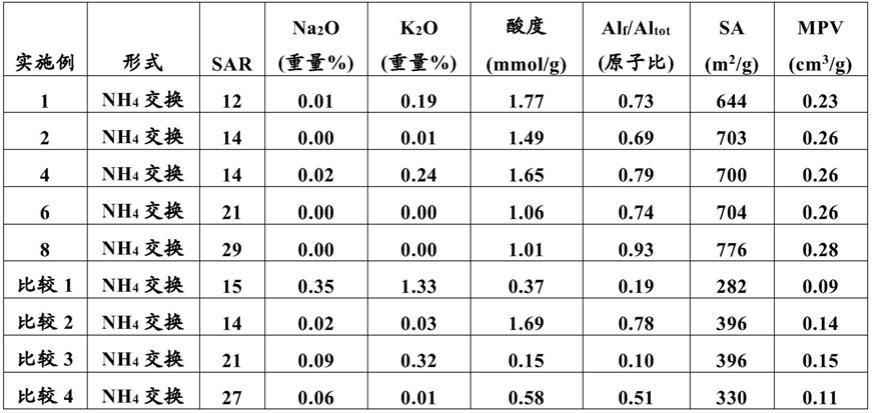
其中r1为衍生或非衍生的c1-c5烷基链,且r2为衍生或非衍生的c2-c5烷基链;并且其中x为h(氢)或者一个或多个衍生或非衍生的c1-c3烷基取代基,所述c1-c3烷基取代基连接至构成六氢-1h-氮杂卓鎓环的碳原子的任何组合。
[0072]
在一个实施方案中,本文所述的季铵阳离子材料以至少0.01重量%、诸如0.01至30重量%、诸如0.01至25重量%、0.1重量%至22%、或1.0至20重量%的量存在。在这些范围中的任何版本也是可能的,诸如0.01至22重量%、0.1至20重量%或0.01至1.0重量%。
[0073]
在一个实施方案中,第一osda可以氢氧化物形式或盐形式使用,盐形式包括但不限于氟化物、氯化物、溴化物、碘化物或乙酸盐形式,或它们的混合物。
[0074]
在一个实施方案中,微孔晶体材料是使用一种或多种osda生产的,其中第二osda是呈氢氧化物形式或盐形式的n,n,n-三甲基-1-金刚烷基铵、n-乙基-n,n-二甲基环己基铵或苄基三甲基铵,盐形式包括但不限于氟化物、氯化物、溴化物、碘化物或乙酸盐形式,或它们的混合物。
[0075]
在另一个实施方案中,第二有机结构导向剂可包含能够形成具有菱沸石(cha)结构的沸石的化合物。例如,第二有机结构导向剂可包含能够形成具有菱沸石(cha)结构的沸石的化合物,诸如胺、单季铵化合物或二季铵化合物。能够形成具有cha结构的沸石的化合物的非限制性实例包括n,n-二甲基-n-乙基环己基铵,n,n-二甲基吡咯烷鎓,n,n-二甲基哌啶鎓,n,n-二甲基六氢氮杂卓鎓,苄基三甲基铵,和它们的混合物。这些化合物、它们的制造方法和使用它们合成cha沸石材料的方法描述于以引用方式并入本文的美国专利号7,670,589、美国专利号7,597,874b1和wo 2013/035054中。
[0076]
在实施方案中,含碱金属的添加剂包含钾、钠或钠和钾的混合物的来源。实例分别包括氢氧化钾,铝酸钾,氢氧化钠和铝酸钠。
[0077]
在实施方案中,铝的来源包括但不限于铝酸钠、铝盐、氢氧化铝、含铝沸石、铝醇盐或氧化铝。二氧化硅的来源可包括但不限于硅酸钠、硅酸钾、硅胶、硅溶胶、气相法二氧化硅、二氧化硅-氧化铝、沸石、硅醇盐或沉淀二氧化硅。
[0078]
在实施方案中,在高压釜中,在120-200℃的温度下加热凝胶1-100小时,诸如180℃、48小时。所述方法可进一步包括过滤凝胶以形成固体产物,用去离子水冲洗固体产物,干燥已冲洗的产物,煅烧干燥的产物,对煅烧产物进行铵或质子交换。
[0079]
测量技术:
[0080]
表面积测量.表面积是根据熟知的bet(brunauer-emmett-teller)氮吸附技术、也被称为“bet法”测定的。本文中在将bet法应用于根据本发明的材料中,遵循astm d4365-95的通用程序和指南。为了确保待测量样品的状态一致,对所有样品进行预处理。适合地,预处理涉及将样品加热至例如400至500℃的温度持续足以清除游离水的时间、诸如3至5小时。在一个实施方案中,预处理包括将每个样品加热至500℃持续4小时。在实施方案中,发明性材料的表面积为500至900m2/g,诸如550至900m2/g,或甚至600至900m2/g。
[0081]
微孔体积测量.微孔体积的评价尤其来源于bet测量技术,利用文献(journal of catalysis 3,32(1964))中所描述的称为t图法(或有时仅被称为t法)的评估方法。
[0082]
在实施方案中,本文所述的沸石类菱沸石材料通常具有0.12cm3/g以上的微孔体积。在实施方案中,发明性材料的微孔体积为0.12至0.30cm3/g,诸如0.15至0.30cm3/g,或甚至0.18至0.30cm3/g。
[0083]
酸度测量.正丙胺被用作探针分子以测定cha材料的酸度,因为正丙胺选择性化学吸附于cha的bronsted酸位点上。热重分析仪(tga)系统用于测量,其中通过加热至280℃去除物理吸附的正丙胺,并且由在280-500℃温度范围内的重量变化测定化学吸附的正丙胺。酸度(酸位点密度)值由280与500℃之间的重量变化计算,单位为mmol/g。d.parrillo等人,applied catalysis,第67卷,第107-118页,1990年的参考文献因其涉及酸度测量的教导而以引用方式并入。
实施例
[0084]
意图为示例性的以下非限制性实施例进一步阐明本发明。
[0085]
实施例1.合成12sar cha
[0086]
将6.1克去离子(di)水、1.7克n,n,n-三甲基-1-金刚烷基氢氧化铵(sachem,25重量%溶液)、8.8克dehhaoh(n,n-二乙基六氢-1h-氮杂卓鎓氢氧化物)(20重量%溶液)、0.53克氢氧化钾(50重量%溶液)添加到一起形成混合物。接下来,将10.1克硅溶胶(ludox hs-40,w.r.grace,40重量%sio2)加入混合物。然后,将2.0克铝酸钠(southern ionics,23.5重量%al2o3)加入混合物,之后是添加0.78克硫酸(macron,97重量%)。接下来,添加0.25克初合成菱沸石粉(14sar)作为晶种。凝胶的摩尔组成为[14.5sio2:1:0al2o3:0.51k2o:1.45na2o:0.44tmaaoh:2.2dehhaoh:261h2o]。在不锈钢高压釜(parr instruments,45ml)中,伴随30rpm下旋转,使所得凝胶在160℃下结晶48小时。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。实施例1的xrd图案示于图1中。根据图1中的xrd图案,实
施例1的样品是相纯菱沸石。
[0087]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有12的sar,0.01重量%的na2o和0.19重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为1.77mmol/g。铵交换样品展现表1中概述的性质。
[0088]
实施例2.合成14sar cha
[0089]
将504克di水、53.4克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、76.7克1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物(sachem,20.4重量%水溶液)、12.2克氢氧化钾(50重量%水溶液)、28.9克硝酸(69重量%水溶液)、0.15克具有cha结构的晶种、271克硅溶胶(nalco,40重量%)和53.9克铝酸钠溶液(southern ionics,23.5重量%al2o3)按此顺序混合到一起。凝胶的摩尔组成为[14.5sio2:1:0al2o3:2.54hno3:0.44k2o:1.52na2o:0.51tmaaoh:0.73dehhaoh:363h2o]。在2l parr高压釜中,伴随150rpm搅动,使用140℃持续24小时之后是180℃持续24小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。实施例2的xrd图案示于图2中。根据图2中的xrd图案,实施例2的样品是相纯菱沸石。根据对实施例2的初合成样品的液相色谱分析,初合成样品含有5.5重量%1,1-二乙基六氢-1h-氮杂卓鎓osda和9.6重量%n,n,n-三甲基金刚烷基铵osda。
[0090]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有14的sar,0.00重量%的na2o和0.01重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为1.49mmol/g。铵交换样品展现表1中概述的性质。
[0091]
实施例3.实施例2的cu交换
[0092]
实施例2的铵交换沸石与硝酸铜进行cu交换以实现3.7重量%cuo的cuo含量。在10%h2o/空气中,将这种cu交换材料在800℃下进一步汽蒸16小时。
[0093]
实施例4.合成14sar cha
[0094]
将756克di水、80.3克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、115.6克1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物(sachem,20.4重量%水溶液)、18.2克氢氧化钾(50重量%水溶液)、43.3克硝酸(69重量%水溶液)、0.20克具有cha结构的晶种、407克硅溶胶(nalco,40重量%)和80.8克铝酸钠溶液(southern ionics,23.5重量%al2o3)按此顺序混合到一起。凝胶的摩尔组成为[14.5sio2:1:0al2o3:2.54hno3:0.44k2o:1.52na2o:0.51tmaaoh:0.73dehhaoh:363h2o]。在2l parr高压釜中,伴随150rpm搅动,使用140℃持续24小时之后是180℃持续24小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。实施例4的xrd图案示于图3中。根据图3中的xrd图案,实施例4的样品是相纯菱沸石。根据对实施例4的初合成样品的液相色谱分析,初合成样品含有5.7重量%1,1-二乙基六氢-1h-氮杂卓鎓osda和9.9重量%n,n,n-三甲基金刚烷基铵osda。
[0095]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有14的sar,0.02重量%的na2o和0.24重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为
1.65mmol/g。铵交换样品展现表1中概述的性质。
[0096]
实施例5.实施例4的cu交换
[0097]
实施例4的铵交换沸石与硝酸铜进行cu交换以实现6.0重量%cuo的cuo含量。在10%h2o/空气中,将这种cu交换材料在750℃下进一步汽蒸16小时。
[0098]
实施例6.合成21sar cha
[0099]
将794克di水、81.4克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、117克1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物(sachem,20.4重量%水溶液)、7.3克固体氢氧化钾、0.5克氢氧化钠(50重量%水溶液)、31.4克硝酸(69重量%水溶液)、1.1克具有cha结构的晶种、55.4克铝酸钠溶液(southern ionics,23.5重量%al2o3)和412克硅溶胶(nalco,40重量%sio2)按此顺序混合到一起。凝胶的摩尔组成为[21sio2:1:0al2o3:2.63hno3:0.42k2o:1.58na2o:0.74tmaaoh:1.05dehhaoh:525h2o]。在2l parr高压釜中,伴随150rpm搅动,使用160℃48小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。实施例6的xrd图案示于图4中。根据图4中的xrd图案,实施例6的样品是相纯菱沸石。根据对实施例6的初合成样品的液相色谱分析,初合成样品含有10重量%1,1-二乙基六氢-1h-氮杂卓鎓osda和10重量%n,n,n-三甲基金刚烷基铵osda。
[0100]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有21的sar,0.00重量%的na2o和0.00重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为1.06mmol/g。铵交换样品展现表1中概述的性质。
[0101]
实施例7.实施例6的cu交换
[0102]
实施例6的铵交换沸石与硝酸铜进行cu交换以实现4.4重量%cuo的cuo含量。在10%h2o/空气中,将这种cu交换材料在800℃下进一步汽蒸16小时。
[0103]
实施例8.合成29sar cha
[0104]
将94.5克di水、80.9克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、203克1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物(sachem,20.4重量%水溶液)、3.5克氢氧化钠(50重量%水溶液)、24.0克硝酸(69重量%水溶液)、1.8克具有cha结构的晶种、359克硅溶胶(nalco,40重量%sio2)和34.6克铝酸钠溶液(southern ionics,23.5重量%al2o3)按此顺序混合到一起。凝胶的摩尔组成为[28sio2:1:0al2o3:3.08hno3:1.82na2o:1.12tmaaoh:2.8dehhaoh:364h2o]。在2l parr高压釜中,伴随150rpm搅动,使用140℃持续24小时之后是180℃持续24小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。实施例8的xrd图案示于图5中。根据图5中的xrd图案,实施例8的样品是相纯菱沸石。根据对实施例8的初合成样品的液相色谱分析,初合成样品含有11重量%1,1-二乙基六氢-1h-氮杂卓鎓osda和9.7重量%n,n,n-三甲基金刚烷基铵osda。
[0105]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有28.8的sar,0.00重量%的na2o和0.00重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为1.01mmol/g。铵交换样品展现表1中概述的性质。
[0106]
实施例9.实施例8的cu交换
[0107]
实施例8的铵交换沸石与硝酸铜进行cu交换以实现4.0重量%cuo的cuo含量。在10%h2o/空气中,将这种cu交换材料在800℃下进一步汽蒸16小时。
[0108]
比较实施例1.合成cha
[0109]
按照实施例1的凝胶配方制备样品,不同之处在于配方中不使用1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物。添加14.2克di水、1.9克n,n,n-三甲基-1-金刚烷基氢氧化铵(sachem,25重量%溶液)、0.91克氢氧化钾(50重量%溶液)、0.44克氢氧化钠溶液(southern ionics,50重量%)以形成混合物。然后,将11.3克硅溶胶(ludox hs-40,w.r.grace,40重量%sio2)加入混合物。接下来,将2.3克铝酸钠(southern ionics,23.5重量%al2o3)加入混合物,之后是0.30克硫酸(macron,97重量%)。然后,添加0.28克初合成菱沸石粉(14sar)作为晶种。凝胶的摩尔组成为[14.5sio2:1:0al2o3:0.78k2o:2.1na2o:0.44tmaaoh:261h2o]。在不锈钢高压釜(parr instruments,45ml)中,伴随30rpm下旋转,使所得凝胶在160℃下结晶48小时。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。比较实施例1的xrd图案示于图6中。根据图6中的xrd图案,比较实施例1的样品是菱沸石和不定形相的混合物。
[0110]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有15的sar,0.31重量%的na2o和1.19重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为0.37mmol/g。铵交换样品展现表1中概述的性质。
[0111]
比较实施例2
[0112]
按照实施例2的凝胶配方制备样品,不同之处在于配方中不使用1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物。将580克di水、54.6克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、12.4克氢氧化钾(50重量%水溶液)、21.1克硝酸(69重量%水溶液)、0.15克具有cha结构的晶种、277克硅溶胶(nalco,40重量%sio2的水中悬浮液)和55.1克铝酸钠溶液(southern ionics,23.5重量%al2o3)按此顺序混合到一起。凝胶的摩尔组成为[14.5sio2:1:0al2o3:1.81hno3:0.44k2o:1.52na2o:0.51tmaaoh:363h2o]。在2l parr高压釜中,伴随150rpm搅动,使用140℃持续24小时之后是180℃持续24小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。比较实施例2的xrd图案示于图7中。根据图7中的xrd图案,比较实施例2的样品是菱沸石和未知相的混合物。
[0113]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有13.6的sar,0.02重量%的na2o和0.03重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为1.69mmol/g。铵交换样品展现表1中概述的性质。
[0114]
比较实施例3
[0115]
按照实施例6的凝胶配方制备样品,不同之处在于配方中不使用1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物。
[0116]
将911克di水、83.2克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、7.4克固体氢氧化钾、0.5克氢氧化钠(50重量%水溶液)、19.3克硝酸(69重量%水溶
液)、1.1克具有cha结构的晶种、56.6克铝酸钠溶液(southern ionics,23.5重量%al2o3)和422克硅溶胶(nalco,40重量%sio2)按此顺序混合到一起。凝胶的摩尔组成为[21sio2:1:0al2o3:1.58hno3:0.42k2o:1.58na2o:0.74tmaaoh:525h2o]。在2l parr高压釜中,伴随150rpm搅动,使用160℃48小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。比较实施例3的xrd图案示于图8中。根据图8中的xrd图案,比较实施例3的样品是菱沸石和不定形相的混合物。
[0117]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有20.5的sar,0.09重量%的na2o和0.32重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为0.15mmol/g。铵交换样品展现表1中概述的性质。
[0118]
比较实施例4
[0119]
按照实施例8的凝胶配方制备样品,不同之处在于配方中不使用1,1-二乙基六氢-1h-氮杂卓鎓氢氧化物。
[0120]
将283克di水、87.0克n,n,n-三甲基金刚烷基氢氧化铵(sachem,25重量%水溶液)、3.7克氢氧化钠(50重量%水溶液)、2.4克硝酸(69重量%水溶液)、2.0克具有cha结构的晶种、386克硅溶胶(nalco,40重量%sio2)和37.3克铝酸钠溶液(southern ionics,23.5重量%al2o3)按此顺序混合到一起。凝胶的摩尔组成为[28sio2:1:0al2o3:0.28hno3:1.82na2o:1.12tmaaoh:364h2o]。在2l parr高压釜中,伴随150rpm搅动,使用140℃持续24小时之后是180℃持续24小时的结晶过程使所得凝胶结晶。将回收的固体过滤,用di水冲洗,并且在105℃下在空气中过夜干燥。比较实施例4的xrd图案示于图9中。根据图9中的xrd图案,比较实施例4的样品是菱沸石和不定形相的混合物。
[0121]
将干燥的沸石粉在空气中在450℃下煅烧1小时,之后是使用3℃/min的升温速率在550℃下6小时。煅烧后,将样品与硝酸铵溶液进行铵交换。在铵交换后,样品具有27.3的sar,0.06重量%的na2o和0.01重量%的k2o。由正丙胺吸附测定的铵交换样品的酸度为0.58mmol/g。铵交换样品展现表1中概述的性质。
[0122]
表1.发明和比较实施例中制备的材料的分析数据
[0123][0124]
铵交换沸石与硝酸铜进行cu交换以实现4-6重量%cuo的cuo含量。在10%h2o/空气中,将cu交换材料在750℃或800℃下进一步汽蒸16小时。在水热处理之前和之后测量cu
交换材料的xrd图案以获得xrd保持率。结果概述于表2中。还评估了已汽蒸cu交换材料的scr活性,并且结果概述于表3中。
[0125]
表2.发明实施例中制备的材料的分析数据
[0126]
实施例蒸汽煅烧温度(℃)sarcuo(重量%)xrd保持率(%)3800143.7725750146.0807800204.4889800264.083
[0127]
表3.发明实施例中制备的材料的分析和scr数据
[0128][0129]
除非有另外指示,否则说明书和权利要求书中使用的表示成分的量、反应条件等等的所有数值应被理解为在所有情况下被术语“约”修饰。因此,除非有相反的指示,否则说明书和随附权利要求书中阐述的数值参数是可视试图由本发明获得的期望性质而变化的近似值。
[0130]
本领域技术人员将通过考虑说明书和本文所公开的发明实践而明白本发明的其他实施方案。预期说明书和实施例被认为仅是示例性的,本发明的真正范围由以下权利要求书指示。