1.本发明涉及双光子材料技术领域,尤其涉及一种含氮化合物及其制备方法和应用。
背景技术:
2.1931年,波尔的学生maria goeppert-mayer在她的毕业论文《双量子跃迁的基元物理过程》中,提出了同时吸收两个光子(即双光子吸收:two-photon absorption,以下简称tpa)的可能性。双光子吸收是指在强激光作用下一个分子同时吸收两个光子的过程,是一种强激光作用下光与物质相互作用的现象。双光子吸收过程中,分子吸收第一个光子到达虚拟态,这个虚拟态并非真实稳定的状态,而是介于基态和激发态之间的一切有可能的状态的组合。由于基态和激发态能量δe差较大,根据海森堡测不准原理,这个虚拟态能够存在的时间很短,只有飞秒量级。直到1961年,kaiser跟garret才首先在caf2:eu
2+
晶体中观察到了双光子吸收现象。当他们用聚焦的红宝石激光器发出的694.3nm的激光束来泵浦caf2:eu
2+
晶体时,观察到了蓝荧光,即能量上转换的荧光,从而第一次在实验上证明了双光子吸收现象的存在。
3.由于发生双光子吸收而引发的光聚合过程称为双光子聚合。双光子聚合微加工是双光子聚合的一个重要应用,双光子聚合微加工可以用来加工一些纳米级的微结构,来用作组织工程支架。用双光子聚合加工的微结构具有更高的分辨率,能够突破衍射极限,使加工的微结构更加细致完整。因此双光子引发剂的效率影响聚合的效果以及适用的激光强度和扫描速度。同时几乎直接决定了制造分辨率。
4.然而,目前的双光子聚合引发剂仍然相对低效并且不足以用于大规模的增材制造。因此,高效双光子聚合引发剂的研究极为重要。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提出一种含氮化合物,能够有效提高双光子聚合反应的引发效率。
6.同时,本发明还提供所述含氮化合物的制备方法和应用。
7.具体地,本发明采取如下的技术方案:
8.本发明的第一方面是提供一种具有如下式i所示结构式的含氮化合物:
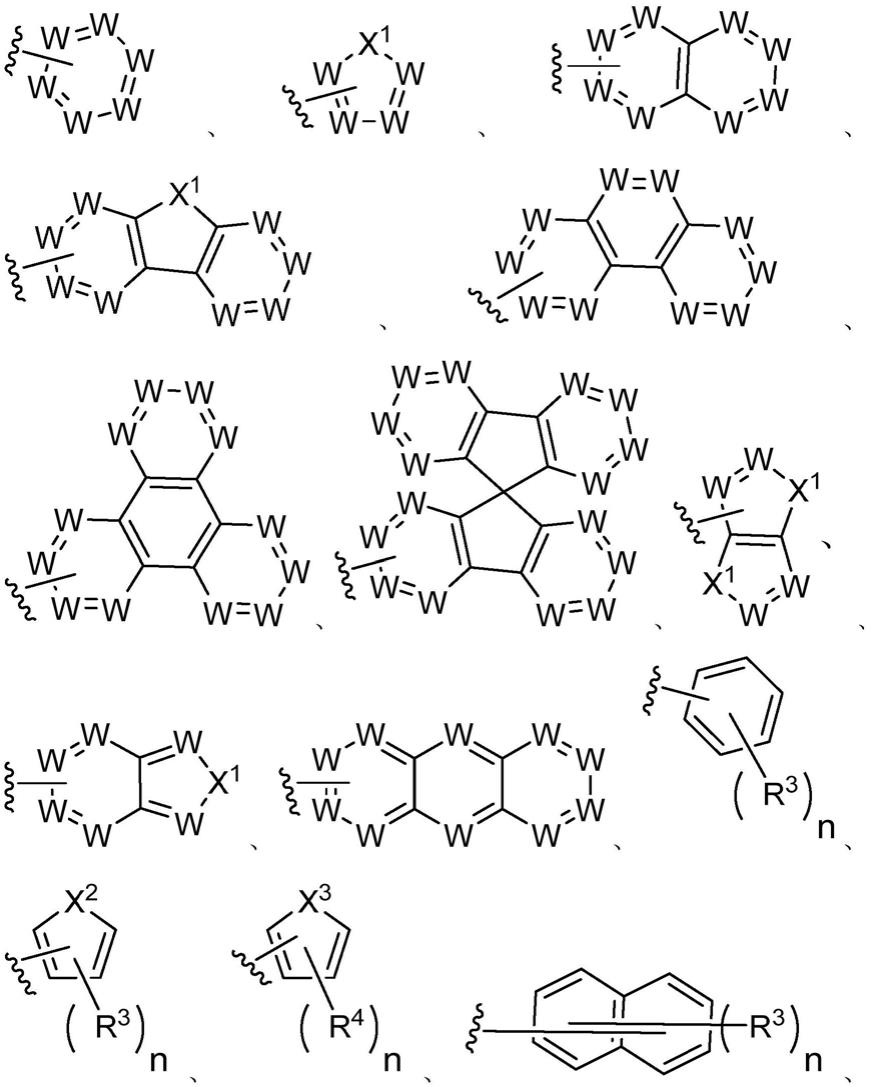
9.10.其中,x选自cr
12
、c=o、cr1=cr1、o或s,或不存在;
11.每个y相互独立地选自n或cr2;
12.每个r2、每个r1相互独立地选自h、卤素、硝基、氰基、异氰基、醛基、具有2至10个c原子的取代或未取代羰基、具有1至10个c原子的取代或未取代酯基、具有1至10个c原子的取代或未取代异氰酸酯基、具有1至10个c原子的取代或未取代烷基、具有2至10个c原子的取代或未取代烯基、具有2至10个c原子的取代或未取代炔基、具有5至61个环原子的取代或未取代芳香基团、具有5至61个环原子的取代或未取代杂芳香基团,并且相邻的基团可以相互成环;
13.r选自具有1至10个c原子的取代或未取代羰基、具有1至10个c原子的取代或未取代酯基、具有1至10个c原子的取代或未取代异氰酸酯基、具有1至10个c原子的取代或未取代烷基、具有2至10个c原子的取代或未取代烯基、具有2至10个c原子的取代或未取代炔基、具有5至61个环原子的取代或未取代芳香基团、具有5至61个环原子的取代或未取代杂芳香基团,或不存在;
14.每个ar相互独立地选自具有5至61个环原子的取代或未取代芳香基团、具有5至61个环原子的取代或未取代杂芳香基团、具有2至10个c原子的取代或未取代烯基、具有2至10个c原子的炔基;
15.每个a相互独立地选自具有5至61个环原子的取代或未取代芳香基团、具有5至61个环原子的取代或未取代杂芳香基团,并且至少一个a为具有拉电子性质的化学结构。
16.在本发明中,“取代”表示被取代基中的氢原子被取代基所取代。
17.相邻的基团指的是邻近基团,即相隔一至两个原子的基团。
[0018]“环原子数”表示原子键合成环状而得到的结构化合物(例如,单环化合物、稠环化合物、交联化合物、碳环化合物、杂环化合物)的构成该环自身的原子之中的原子数。该环被取代基所取代时,取代基所包含的原子不包括在成环原子内。关于以下所述的“环原子数”,在没有特别说明的条件下也是同样的。例如,苯环的环原子数为6,萘环的环原子数为10,噻吩基的环原子数为5。
[0019]
芳香基团指至少包含一个芳环的烃基,包括单环基团和多环的环系统。杂芳香基团指包含至少一个杂芳香环的烃基(含有杂原子),包括单环基团和多环的环系统。杂原子优选选自si、n、p、o、s、ge中的至少一种,特别优选选自si、n、p、o、s中的至少一种。这些多环的环可以具有两个或多个环,其中两个碳原子被两个相邻的环共用,即形成稠环。多环的这些环中,至少一个是芳族的或杂芳族的。对于本发明的目的,芳香基团或杂芳香基团不仅包括芳香基或杂芳香基的体系,而且,其中多个芳基或杂芳香基也可以被短的非芳族单元间断(《10%的非h原子,优选小于5%的非h原子,比如c、n或o原子)。因此,比如9,9'-螺二芴,9,9-二芳基芴,三芳胺,二芳基醚等体系,对于该发明目的同样认为是芳香族基团。
[0020]
具体地,芳香基团的例子有:苯、联苯、萘、蒽、菲、苯并菲、二萘嵌苯、并四苯、芘、苯并芘、苝、三亚苯、苊、芴、三苯胺、三苯基氧磷、四苯基硅,及它们的衍生物,优选苯、联苯、萘、蒽、菲、苯并菲、芘、苝、三苯胺、三苯基氧磷、四苯基硅中的至少一种。
[0021]
具体地,杂芳香基团的例子有:呋喃、苯并呋喃、噻吩、苯并噻吩、吡咯、吡唑、三唑、咪唑、噁唑、噁二唑、噻唑、四唑、吲哚、咔唑、吡咯并咪唑、吡咯并吡咯、噻吩并吡咯、噻吩并噻吩、呋喃并吡咯、呋喃并呋喃、噻吩并呋喃、苯并异噁唑、苯并异噻唑、苯并咪唑、吡啶、吡
嗪、哒嗪、嘧啶、三嗪、喹啉、异喹啉、邻二氮萘、喹喔啉、菲啶、伯啶、喹唑啉、喹唑啉酮、硫芴、硅芴、螺芴、螺硅芴,及它们的衍生物,优选吡啶、嘧啶、三嗪、硫芴、硅芴、螺芴、螺硅芴、咔唑、噻吩、呋喃、噻唑、噁二唑中的至少一种。
[0022]
根据本发明第一方面的含氮化合物,至少具有如下有益效果:
[0023]
本发明的含氮化合物具有多环结构,具有很好的双光子吸收截面,能够有效提高双光子聚合反应的引发效率。
[0024]
在发明的一些实施方式中,每个a相互独立地选自如下任一种基团:
[0025][0026]
其中,每个w独立地选自n、cr5、c=o,并且最少有一个w为n或者c=o;
[0027]
每个x1相互独立地选自cr
62
、c=o、o、s、s=o、s=(o)2;
[0028]
每个x2相互独立地选自cr
72
、o、s;
[0029]
每个x3相互独立地选自c=o、s=o、s=(o)2;
[0030]
每个r3~r7相互独立地选自h、卤素、硝基、氰基、异氰基、醛基、具有2至10个c原子的取代或未取代羰基、具有1至10个c原子的取代或未取代酯基、具有1至10个c原子的取代或未取代异氰酸酯基、具有1至10个c原子的取代或未取代烷基、具有2至10个c原子的取代或未取代烯基、具有2至10个c原子的取代或未取代炔基、具有5至61个环原子的取代或未取代芳香基团、具有5至61个环原子的取代或未取代杂芳香基团,并且相邻的两个基团可以相互连接成环;其中最少有一个r3选自卤素、硝基、氰基、异氰基、醛基、具有2至10个c原子的取代或未取代羰基;
[0031]
每个n相互独立地表示0~10的整数。
[0032]
在本发明的一些实施方式中,每个a相互独立地选自如下任一种基团:
[0033][0034]
其中,r3、r4、r5、x1、x2的定义如前所述,m选自0~10的整数。
[0035]
在本发明的一些实施方式中,所述含氮化合物具有如下任意一种结构式:
[0036]
[0037]
[0038][0039]
其中,x、y、r、ar、r3、r4、r5和x1的定义如前所述。
[0040]
在本发明的一些实施方式中,所述含氮化合物具有如下任意一种结构式:
[0041]
[0042]
[0043]
[0044]
[0045]
[0046]
[0047][0048]
在本发明的一些实施方式中,在25℃下,所述含氮化合物在甲苯中的溶解度≥0.05mg/ml,优选≥2mg/ml,最优选≥5mg/ml;在25℃下,所述含氮化合物在水中的溶解度≥0.01mg/ml,优选≥0.1mg/ml,最优选≥1mg/ml。
[0049]
本发明的第二方面是提供所述含氮化合物的制备方法,当存在r时,包括如下步骤:
[0050]
使化合物1-1a-b(oh)2与化合物1-2反应,得到化合物1-3其中m表示卤素;
[0051]
对所述化合物1-3中的r进行卤素取代,得到化合物1-4
[0052]
使所述化合物1-4与化合物1-5反应,得到含氮化合物
[0053]
所述基团x、y、r、ar、a如前所述,且r是存在的。
[0054]
在本发明的一些实施方式中,所述化合物1-1与化合物1-2的摩尔比为2~2.5:1,优选约2:1。所述化合物1-1与化合物1-2之间的反应是一种交叉偶联反应,在反应过程中可在本领域通用的偶联反应用催化剂作用下进行,作为示例,所述催化剂可采用钯配合物,例如pd(pph3)4。且,所述化合物1-1与化合物1-2的反应在碱性条件下进行,在实际操作中,可通过在反应体系中加入碳酸钠、氢氧化钠、碳酸钾、氢氧化钾等碱性物质形成碱性的反应条件,所述碱性物质与化合物1-2的摩尔比可设为3~5:1,也可以根据实际情况选用其他比例的碱性物质。同时,化合物1-1与化合物1-2反应所采用的溶剂可根据实际情况选择本领域通用的溶剂,例如选用甲苯、二氯甲烷、水、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺中的任意一种或任意多种的混合。所述化合物1-1与化合物1-2优选在甲苯与水的混合溶剂中进行反应,所述甲苯与水的体积比可设为2~4:1;反应物与溶剂的比例可根据实际情况进行设置,例如可将溶剂与化合物1-1的比例设为10~20ml:1mmol。
[0055]
在本发明的一些实施方式中,所述化合物1-1与化合物1-2的反应温度为70~100℃,优选80~90℃,更优选约85℃。所述反应的时间为5~15h。所述反应在惰性保护氛围下进行。
[0056]
在本发明的一些实施方式中,可通过使所述化合物1-3与卤素单质混合来进行卤
素取代,所述卤素单质与化合物1-3的摩尔比为0.9~1.2:1。
[0057]
在本发明的一些实施方式中,所述化合物1-3与卤素单质的混合温度为-10~10℃,优选-5~0℃,更有选约0℃,在实际操作中,可在冰浴条件下将二者进行混合。将化合物1-3与卤素单质混合完毕之后,将温度升至0~40℃,优选20~30℃进行卤素取代,所述卤素取代的时间为5~15h。达到所需反应时间后,可通过在反应体系中加入亚硫酸盐溶液来终止反应。
[0058]
在本发明的一些实施方式中,所述化合物1-4与化合物1-5的摩尔比为0.8~1.2:1,优选约1:1。所述化合物1-4与化合物1-5的反应过程可在钯配合物[如pd(dba)2、t-bu3p、pd(pph3)4等]和/或醇盐(如naobu-t)等的催化作用下进行。在一些优选的实施例中,所述催化剂与化合物1-4的摩尔比为1~3:1;在一些更优选地实施例中,所述催化剂为pd(dba)2、t-bu3p和naobu-t三者的组合,所述pd(dba)2、t-bu3p和naobu-t的摩尔比为1:(2~3):(150~250)。同时,所述化合物1-4与化合物1-5反应所采用的溶剂可根据实际情况选择本领域通用给无水有机溶剂,例如无水甲苯等。
[0059]
在本发明的一些实施方式中,所述化合物1-4与化合物1-5的反应温度为80~100℃,优选85~95℃;反应的时间为2~5h。
[0060]
或者,存在r时,所述含氮化合物的制备方法包括如下步骤:
[0061]
使化合物2-1与化合物2-2反应,得到化合物2-3其中m表示卤素;
[0062]
对所述化合物2-3进行卤素取代,得到化合物2-4
[0063]
使所述化合物2-4与化合物2-5a-h反应,得到含氮化合物
[0064]
所述基团x、y、r、ar、a如前所述,且r是存在的。
[0065]
在本发明的一些实施方式中,所述化合物2-1与化合物2-2的摩尔比为0.8~1.2:1,优选约1:1。所述化合物2-1与化合物2-2的反应可在本领域通用的偶联反应用催化剂作用下进行,作为示例,所述催化剂可采用钯配合物,例如pd(pph3)4。且,所述化合物2-1与化合物2-2的反应在碱性条件下进行,在实际操作中,可通过在反应体系中加入碳酸钠、氢氧化钠、碳酸钾、氢氧化钾等碱性物质形成碱性的反应条件,所述碱性物质与化合物2-2的摩尔比可设为3~5:1,也可以根据实际情况选用其他比例的碱性物质。同时,化合物2-1与化合物2-2反应所采用的溶剂可根据实际情况选择本领域通用的溶剂,例如选用甲苯、二氯甲烷、水、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺中的任意一种或任意多种的混合。所述化合物2-1与化合物2-2优选在甲苯与水的混合溶剂中进行反应,所述甲苯与水的体积比可设为2~4:1;反应物与溶剂的比例可根据实际情况进行设置,例如可将溶剂与化合物2-1的比例设为10~20ml:1mmol。
[0066]
在本发明的一些实施方式中,所述化合物2-1与化合物2-2的反应温度为70~100℃,优选80~90℃,更优选约85℃。所述反应的时间为5~15h。所述反应在惰性保护氛围下进行。
[0067]
在本发明的一些实施方式中,可通过使所述化合物2-3与卤素单质混合来进行卤素取代,所述卤素单质与化合物2-3的摩尔比为2~2.5:1。
[0068]
在本发明的一些实施方式中,所述化合物2-3与卤素单质的混合温度为-10~10℃,优选-5~0℃,更有选约0℃,在实际操作中,可在冰浴条件下将二者进行混合。将化合物2-3与卤素单质混合完毕之后,将温度升至0~40℃,优选20~30℃进行卤素取代,所述卤素取代的时间为5~15h。达到所需反应时间后,可通过在反应体系中加入亚硫酸盐溶液来终止反应。
[0069]
在本发明的一些实施方式中,所述化合物2-5与化合物2-4的摩尔比为2~5:1,优选4~5:1。所述化合物2-5与化合物2-4的反应过程可在钯配合物[如醋酸钯等]的催化作用下进行。在一些优选的实施例中,所述催化剂与化合物2-4的比例为150~250mg:1mmol。优选地,所述化合物2-5与化合物2-4的反应过程在有机还原剂(如三甲苯基膦)作用下进行,所述有机还原剂与化合物2-4的比例为0.5~1g:1mmol。优选地,所述化合物2-5与化合物2-4的反应体系中还可加入有机胺(如三乙胺),所述有机胺与化合物2-4的比例为50~80ml:1mmol。
[0070]
在本发明的一些实施方式中,所述化合物2-5与化合物2-4的反应温度为100~150℃,优选100~120℃;反应的时间为20~36h。
[0071]
或者,当r不存在时,所述含氮化合物的制备方法包括如下步骤:
[0072]
使化合物3-1a-b(oh)2与化合物3-2反应,得到化合物3-3其中m表述卤素;
[0073]
使所述化合物3-3与化合物3-4反应,得到含氮化合物
[0074]
所述基团x、y、ar、a如前所述。
[0075]
在本发明的一些实施方式中,所述化合物3-1与化合物3-2的摩尔比为2~2.5:1,优选约2:1。所述化合物3-1与化合物3-2之间的反应过程中可在本领域通用的偶联反应用催化剂作用下进行,作为示例,所述催化剂可采用钯配合物,例如pd(pph3)4。且,所述化合物3-1与化合物3-2的反应在碱性条件下进行,在实际操作中,可通过在反应体系中加入碳酸钠、氢氧化钠、碳酸钾、氢氧化钾等碱性物质形成碱性的反应条件,所述碱性物质与化合物3-2的摩尔比可设为3~5:1,也可以根据实际情况选用其他比例的碱性物质。同时,化合物3-1与化合物3-2反应所采用的溶剂可根据实际情况选择本领域通用的溶剂,例如选用甲苯、二氯甲烷、水、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺中的任意一种或任意多种的混合。所述化合物3-1与化合物3-2优选在甲苯与水的混合溶剂中进行反应,所述甲苯与水的体积比可设为2~4:1;反应物与溶剂的比例可根据实际情况进行设置,例如可将溶剂与化合物3-1的比例设为10~20ml:1mmol。
[0076]
在本发明的一些实施方式中,所述化合物3-1与化合物3-2的反应温度为70~100℃,优选80~90℃,更优选约85℃。所述反应的时间为5~15h。所述反应在惰性保护氛围下进行。
[0077]
在本发明的一些实施方式中,所述化合物3-3与化合物3-4的摩尔比为0.8~1.2:1,优选约1:1。所述化合物3-3与化合物3-4的反应过程可在钯配合物[如pd(dba)2、t-bu3p、pd(pph3)4等]和/或醇盐(如naobu-t)等的催化作用下进行。在一些优选的实施例中,所述催化剂与化合物3-3的摩尔比为1~3:1;在一些更优选地实施例中,所述催化剂为pd(dba)2、t-bu3p和naobu-t三者的组合,所述pd(dba)2、t-bu3p和naobu-t的摩尔比为1:2~3:150~250。同时,所述化合物3-3与化合物3-4反应所采用的溶剂可根据实际情况选择本领域通用
给无水有机溶剂,例如无水甲苯等。
[0078]
在本发明的一些实施方式中,所述化合物3-3与化合物3-4的反应温度为80~100℃,优选85~95℃;反应的时间为2~5h。
[0079]
本发明的第三方面是提供一种组合物,所述组合物的原料含有上述含氮化合物。
[0080]
在本发明的一些实施方式中,所述组合物的原料还包括功能材料,所述功能材料可根据用途进行灵活选择,例如包括光敏树脂材料、聚合单体材料、着色剂材料、助引发剂材料、添加剂材料、可交联高聚物材料、有机染料中的至少一种。所述含氮化合物与功能材料的比例可根据实际情况进行设置,例如将含氮化合物与功能材料的摩尔比设为1:0.00001~100000。
[0081]
当含有聚合物单体材料时,所述含氮化合物可起到引发剂的作用,所述引发剂相较聚合物单体材料的重量百分比≤15wt%,较好是≤12wt%,更好是≤9wt%,更更好是≤8wt%,最好是≤7wt%。
[0082]
所述光敏树脂材料包括丙烯酸树脂和/或丙烯酸酯树脂。
[0083]
所述助引发剂包括脂肪族叔胺、乙醇胺类叔胺、叔胺性苯甲酸酯和活性胺中的至少一种。
[0084]
根据实际需要,所述组合物的原料还可以含有至少一种溶剂,所述溶剂包括水、有机溶剂中的至少一种。所述有机溶剂包括醇类、芳族、杂芳族、酯、芳族酮、芳族醚、脂肪族酮、脂肪族醚、脂环族、烷烃、烯烃类溶剂中的至少一种,其中酯类溶剂包括羧酸酯、硼酸酯、磷酸酯类溶剂中的至少一种;优选包括芳族、杂芳族溶剂中的至少一种。
[0085]
适合本发明的芳族或杂芳族溶剂的例子有,但不限制于:对二异丙基苯、戊苯、四氢萘、环己基苯、氯萘、1,4-二甲基萘、3-异丙基联苯、对甲基异丙苯、二戊苯、三戊苯、戊基甲苯、邻二乙苯、间二乙苯、对二乙苯、1,2,3,4-四甲苯、1,2,3,5-四甲苯、1,2,4,5-四甲苯、丁苯、十二烷基苯、二己基苯、二丁基苯、对二异丙基苯、环己基苯、苄基丁基苯、二甲基萘、3-异丙基联苯、对甲基异丙苯、1-甲基萘、1,2,4-三氯苯、4,4-二氟二苯甲烷、1,2-二甲氧基-4-(1-丙烯基)苯、二苯甲烷、2-苯基吡啶、3-苯基吡啶、n-甲基二苯胺、4-异丙基联苯、α,α-二氯二苯甲烷、4-(3-苯基丙基)吡啶、苯甲酸苄酯、1,1-双(3,4-二甲基苯基)乙烷、2-异丙基萘、喹啉、异喹啉、2-呋喃甲酸甲酯、2-呋喃甲酸乙酯等;
[0086]
适合本发明的芳族酮溶剂的例子有,但不限制于:1-四氢萘酮,2-四氢萘酮,2-(苯基环氧)四氢萘酮,6-(甲氧基)四氢萘酮,苯乙酮、苯丙酮、二苯甲酮、及它们的衍生物,如4-甲基苯乙酮、3-甲基苯乙酮、2-甲基苯乙酮、4-甲基苯丙酮、3-甲基苯丙酮、2-甲基苯丙酮等;
[0087]
适合本发明的芳族醚溶剂的例子有,但不限制于:3-苯氧基甲苯、丁氧基苯、对茴香醛二甲基乙缩醛、四氢-2-苯氧基-2h-吡喃、1,2-二甲氧基-4-(1-丙烯基)苯、1,4-苯并二噁烷、1,3-二丙基苯、2,5-二甲氧基甲苯、4-乙基苯乙醚、1,3-二丙氧基苯、1,2,4-三甲氧基苯、4-(1-丙烯基)-1,2-二甲氧基苯、1,3-二甲氧基苯、缩水甘油基苯基醚、二苄基醚、4-叔丁基茴香醚、反式-对丙烯基茴香醚、1,2-二甲氧基苯、1-甲氧基萘、二苯醚、2-苯氧基甲醚、2-苯氧基四氢呋喃、乙基-2-萘基醚。
[0088]
适合本发明的脂肪族酮溶剂的例子有,但不限制于:2-壬酮、3-壬酮、5-壬酮、2-癸酮、2,5-己二酮、2,6,8-三甲基-4-壬酮、葑酮、佛尔酮、异佛尔酮、二正戊基酮等;或脂肪族
醚,例如,戊醚、己醚、二辛醚、乙二醇二丁醚、二乙二醇二乙醚、二乙二醇丁基甲醚、二乙二醇二丁醚、三乙二醇二甲醚、三乙二醇乙基甲醚、三乙二醇丁基甲醚、三丙二醇二甲醚、四乙二醇二甲醚等。
[0089]
适合本发明的羧酸酯溶剂的例子有,但不限制于:辛酸烷酯、癸二酸烷酯、硬脂酸烷酯、苯甲酸烷酯、苯乙酸烷酯、肉桂酸烷酯、草酸烷酯、马来酸烷酯、烷内酯、油酸烷酯等,特别优选辛酸辛酯、癸二酸二乙酯、邻苯二甲酸二烯丙酯、异壬酸异壬酯。
[0090]
上述所列的溶剂可以是单独使用,也可以是作为两种或多种有机溶剂的混合物使用。
[0091]
在某些优选的实施例中,按照本发明的一种组合物,包含有一种如上所述的含氮化合物或混合物及至少一种有机溶剂,还可进一步包含另一种有机溶剂,另一种有机溶剂的例子,包括但不限于:甲醇、乙醇、2-甲氧基乙醇、二氯甲烷、三氯甲烷、氯苯、邻二氯苯、四氢呋喃、苯甲醚、吗啉、甲苯、邻二甲苯、间二甲苯、对二甲苯、1,4二氧杂环己烷、丙酮、甲基乙基酮、1,2二氯乙烷、3-苯氧基甲苯、1,1,1-三氯乙烷、1,1,2,2-四氯乙烷、醋酸乙酯、醋酸丁酯、二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、四氢萘、萘烷、茚和/或它们的混合物。
[0092]
按照本发明的组合物,其中有机溶剂在选取时需考虑其沸点参数。本发明中,所述的有机溶剂的沸点≥80℃;优选为≥130℃;较优选为≥150℃;最优为≥200℃。
[0093]
本发明的第四方面是提供一种3d打印墨水,所述3d打印墨水的原料含有所述组合物。
[0094]
本发明还提供所述含氮化合物在制作荧光材料或光固化材料中的应用。
[0095]
本发明还提供一种有机电子器件,制备所述有机电子器件的原料含有上述含氮化合物,或制备所述有机电子器件的原料含有上述组合物,或所述有机电子器件含有由所述3d打印墨水经3d打印制造技术形成的元件。
[0096]
所述的3d打印制造技术可选于,但不限于,光固化成型(stereo lithography apparatu,sla)、分层实体制造(layered solid manufacturing,lom)、选择性激光烧结成型(selective laser sintering,sls)、熔融沉积式成型(fused deposition modeling,fdm)和双光子3d打印(two-photonpolymerization,tpp)。
[0097]
在本发明的一些实施方式中,所述有机电子器件包括有机发光二极管(oled)、有机光伏电池(opv)、有机发光电池(oleec)、有机场效应管(ofet)、有机发光场效应管、有机激光器、有机自旋电子器件、光信息存储器件、有机传感器、有机等离激元发射二极管(organic plasmon emitting diode)中的至少一种。
[0098]
相对于现有技术,本发明具有如下有益效果:
[0099]
本发明的含氮化合物具有良好的双光子吸收截面,同时能有效地提高聚合反应的引发效率,克服了现有技术存在的双光子聚合引发效率低等问题。
附图说明
[0100]
图1为实施例2的化合物a-2的紫外可见光吸收光谱和荧光光谱图。
具体实施方式
[0101]
以下结合具体的实施例进一步说明本发明的技术方案。以下实施例中所用的原
料,如无特殊说明,均可从常规商业途径得到;所采用的工艺,如无特殊说明,均采用本领域的常规工艺。
[0102]
实施例1
[0103]
化合物a-1的合成
[0104][0105]
(1)中间体3的合成
[0106]
依次将化合物1(20.8mmol),化合物2(10.36mmol),pd(pph3)4(601mg),碳酸钠(34.72mmol),甲苯200ml和水70ml加入到500ml双口反应瓶中,n2保护,85℃反应过夜。点板反应完,降至室温,分液,水相用二氯甲烷dcm萃取两次。合并有机相,减压蒸干,拌硅胶过柱,dcm/pe(石油醚)=1/20(v/v)过出产品,减压蒸干得6.7g中间体3。
[0107]
(2)中间体4的合成
[0108]
冰浴下将中间体3(5mmol)溶于100ml二氯甲烷中,再缓慢滴加1.17ml液溴(5.3mmol)的二氯甲烷溶液,滴完后缓慢回到室温,继续搅拌反应过夜,加入亚硫酸钠水溶液终止反应,有机相用碳酸钠水溶液和水萃取多次,干燥有机相,柱色谱分离后得1.88g中间体4。
[0109]
(3)化合物a-1的合成
[0110]
在1000ml两口瓶中加入中间体4(0.1mol),化合物5(0.1mol),pd(dba)2(0.003mol),t-bu3p(0.009mol),naobu-t(0.2mol),无水甲苯,90℃反应3小时。后处理水洗,无水硫酸镁干燥,石油醚过硅胶柱,得到化合物a-1,收率89%。
[0111]
化合物a-1的核磁共振氢谱和质谱的表征结果为:1h nmr(300mhz,cdcl3):δ9.06(2h,s),8.61(2h,d),8.42(2h,d),8.10(1h,s),7.80
–
7.12(20h,m));esi-ms(m/z):610(m+h)
+
.elemental analyses(c,h,n)anal.calc.for c
41h27
n4os:c,80.76;h,4.46;n,6.89,found:c,81.03;h,4.42;n,6.97。
[0112]
实施例2
[0113]
化合物a-2的合成
[0114][0115]
(1)中间体7的合成
[0116]
依次将化合物6(20mmol),化合物2(10mmol),pd(pph3)4(600mg),碳酸钠(35mmol),甲苯200ml和水70ml加入到500ml双口反应瓶中,n2保护,85℃反应过夜。点板反应完,降至室温,分液,水相用dcm萃取两次。合并有机相,减压蒸干,拌硅胶过柱,dcm/pe=1/20(v/v)过出产品,减压蒸干得8.0g中间体7。
[0117]
(2)中间体8的合成
[0118]
冰浴下将中间体7(5mmol)溶于100ml二氯甲烷中,再缓慢滴加1.17ml液溴(5.3mmol)的二氯甲烷溶液,滴完后缓慢回到室温,继续搅拌反应过夜,加入亚硫酸钠水溶液终止反应,有机相用碳酸钠水溶液和水萃取多次,干燥有机相,柱色谱分离后得3.8g中间体8。
[0119]
(3)化合物a-2的合成
[0120]
在1000ml两口瓶中加入中间体8(0.1mol),化合物9(0.1mol),pd(dba)2(0.003mol),t-bu3p(0.009mol),naobu-t(0.2mol),无水甲苯,90℃反应3小时。后处理水洗,无水硫酸镁干燥,石油醚过硅胶柱,得到化合物a-2,收率62%。
[0121]
化合物a-2的核磁共振氢谱和质谱的表征结果为:1h nmr(300mhz,cdcl3):δ8.52(1h,s),8.13(4h,m),7.90
–
7.48(22h,m),7.37(6h,m),2.65-3.00(4h,m),1.92-2.23(4h,m),1.30(32h,m),0.53(12h,m));esi-ms(m/z):1141(m+h)
+
.elemental analyses(c,h,n)anal.calc.for c
83h85
n3o:c,87.40;h,7.51;n,3.68,found:c,87.43;h,7.50;n,3.65。
[0122]
化合物a-2的紫外可见吸收光谱和荧光光谱图如图1所示,谱图显示,化合物a-2具有紫外可见光吸收性能以及荧光性能。
[0123]
实施例3
[0124]
化合物a-3的合成
[0125][0126]
(1)中间体12的合成
[0127]
依次将化合物10(20mmol),化合物11(20mmol),pd(pph3)4(600mg),碳酸钠(35mmol),甲苯200ml和水70ml加入到500ml双口反应瓶中,n2保护,85℃反应过夜。点板反应完,降至室温,分液,水相用dcm萃取两次。合并有机相,减压蒸干,拌硅胶过柱,dcm/pe=1/20(v/v)过出产品,减压蒸干得5.7g中间体12。
[0128]
(2)中间体13的合成
[0129]
冰浴下将中间体12(2.5mmol)溶于100ml二氯甲烷中,再缓慢滴加1.17ml液溴(5.3mmol)的二氯甲烷溶液,滴完后缓慢回到室温,继续搅拌反应过夜,加入亚硫酸钠水溶液终止反应,有机相用碳酸钠水溶液和水萃取多次,干燥有机相,柱色谱分离后得1.2g中间体13。
[0130]
(3)化合物a-3的合成
[0131]
将中间体13(0.3mmol)、化合物14(1.3mmol)、醋酸钯57mg以及三甲苯基膦0.205g加入反应器中,抽真空换氮气三次后,加入18ml新蒸三乙胺,110℃反应24小时,蒸去三乙胺后,残留固体用1:4(v:v)的二氯甲烷:石油醚为展开剂柱层析得白色固体化合物a-3,收率43%。
[0132]
化合物a-3的核磁共振氢谱和质谱的表征结果为:1h nmr(300mhz,cdcl3):δ8.55(1h,s),8.30-8.28(4h,m),8.05-7.75(8h,m),7.55(2h,d),7.37(2h,d),7.22
–
7.13(6h,m),5.01(4h,m),3.34(4h,m),1.50-1.30(8h,m),0.60(6h,m));esi-ms(m/z):836(m+h)
+
.elemental analyses(c,h,n)anal.calc.for c
54h45
no4s2:c,77.58;h,5.43;n,1.68,found:c,77.53;h,5.43;n,1.68。
[0133]
实施例4
[0134]
化合物a-4的合成
[0135][0136]
(1)中间体17的合成
[0137]
依次将化合物15(20mmol),化合物16(10mmol),pd(pph3)4(600mg),碳酸钠(35mmol),甲苯200ml和水70ml加入到500ml双口反应瓶中,n2保护,85℃反应过夜。点板反应完,降至室温,分液,水相用dcm萃取两次。合并有机相,减压蒸干,拌硅胶过柱,dcm/pe=1/20(v/v)过出产品,减压蒸干得3.5g中间体17。
[0138]
(2)化合物a-4的合成
[0139]
在1000ml两口瓶中加入化合物17(0.1mol),化合物18(0.1mol),pd(dba)2(0.003mol),t-bu3p(0.009mol),naobu-t(0.2mol),无水甲苯为溶剂,90℃反应3小时。后处理水洗,无水硫酸镁干燥,石油醚过硅胶柱,得到化合物a-4,收率89%。
[0140]
化合物a-4的核磁共振氢谱和质谱的表征结果为:1h nmr(300mhz,cdcl3):δ8.32-8.27(4h,m),8.05(2h,d),7.85-7.80(3h,m),7.57-7.30(10h,m),7.00(2h,m),4.45(4h,m),1.75-1.45(8h,m),0.61(6h,m));esi-ms(m/z):674(m+h)
+
.elemental analyses(c,h,n)anal.calc.for c
44h39
n3o4:c,78.43;h,5.83;n,6.24,found:c,78.50;h,5.81;n,6.24。
[0141]
实施例5
[0142]
避光条件下,在装有搅拌子的玻璃容器中加入900mg实施例1的化合物a-1(起引发剂作用),90g乙氧基化三羟甲基丙烷三丙烯酸酯(单体),搅拌振荡24h,使化合物a-1溶解完全。
[0143]
将化合物a-1分别替换为等质量的实施例2~4的化合物a-2、a-3、a-4,或替换为二苯甲酮bp(对比例1),或m2cmk(对比例2),得到不同的混合物。
[0144]
(1)单光子聚合效率
[0145]
测试方法:将上述配制好的混合物滴涂于干净的玻璃片上,制得膜层,90℃下前烘120s,采用365nm紫外光进行曝光,曝光量40mj/cm2,在23℃显影50s(氢氧化钠溶液,oh-浓度为0.5%),230℃后烘20min,将得到的薄膜进行减重测试。
[0146]
减重率=(第一次烘干后膜及玻璃片重量-第二次烘干后膜及玻璃片重量)/第一次烘干后膜及玻璃片重量。
[0147]
结果如表1所示。
[0148]
表1.
[0149][0150][0151]
测试结果显示,以实施例1~4的含氮化合物作为引发剂时,相比常用的引发剂bp或m2cmk,其减重率更小,说明未聚合单体量上更少,具有更好的光引发效果。
[0152]
(2)双光子聚合效率
[0153]
测试方法:将配制好的混合物滴涂于干净的玻璃片上,制得膜层,90℃下前烘120s,在800nm波长的飞秒激光、80fs脉冲的飞秒激光进行曝光,在23℃显影50s(氢氧化钠溶液,oh-浓度为0.5%),230℃后烘20min,将得到的薄膜进行减重测试。
[0154]
减重率=(第一次烘干后膜及玻璃片重量-第二次烘干后膜及玻璃片重量)/第一次烘干后膜及玻璃片重量。
[0155]
结果如表2所示。
[0156]
表2.
[0157] 光引发剂光引发剂:单体(wt)减重率(%)实施例1a-11:1001.4实施例2a-21:1002.1实施例3a-31:1001.5实施例4a-41:1001.5对比例1bp1:10098.0对比例2m2cmk1:1004.3
[0158]
测试结果显示,以实施例1~4的含氮化合物作为双光子聚合引发剂时,相比常用的引发剂bp或m2cmk,其减重率更小,说明在未聚合单体量上更少,具有更好的双光子光引发效果。因此,可根据需要将这些含氮化合物与光敏树脂材料、聚合单体材料、着色剂材料、助引发剂材料、添加剂材料、可交联高聚物材料、有机染料等混合制成一定用途的组合物,例如3d打印墨水,再经过加工制成各种有机电子器件及其元件。
[0159]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。