1.本发明属于除草剂中间体制备领域,具体涉及一种单杂含量低的2,3-二氟-5-氯吡啶的绿色生产工艺。
背景技术:
2.炔草酯是含氟、高效手性芳氧苯氧丙酸酯类除草剂,主要防除小麦、黑麦、黑小麦等谷物田禾本科杂草,尤其在看麦娘和野燕麦等难治杂草的防治上。炔草酯为内吸传导性除草剂,作用机理为经由植物的叶片和叶鞘吸收,韧皮部传导,积累于植物体的分生组织内,抑制乙酰辅酶a羧化酶(accase),使脂肪酸合成停止,细胞的生长分裂不能正常进行,膜系统等含脂结构破坏,最后导致植物死亡。
3.炔草酯对小麦的安全性较好,施药对小麦苗龄的要求不高,除草效果好,特别是对菵草、硬草等已对精噁唑禾草灵产生抗性的杂草有良好防效;其另一个优势是对低温的适应性好,低温环境下施药不会对麦苗造成不良影响,虽然杀草速度会减慢,但不影响最终防效。炔草酯耐雨水冲刷,使用期宽,且对小麦和后茬作物安全等特点。
4.炔草酯在广泛的环境和气候条件下,提供稳定的防效;具有非常广泛的杀草谱,能防除许多重要的禾本科杂草,并对作物安全;作为优秀的配伍产品,适合与许多产品复配,从而扩大防治谱,而且对环境生态安全等特点,符合当前发展趋势,越来越受到广泛关注和重视。
5.根据炔草酯的合成路线,2,3-二氟-5-氯吡啶是其制备过程中的关键中间体,开发2,3-二氟-5-氯吡啶制备方法具有重要意义。
[0006][0007]
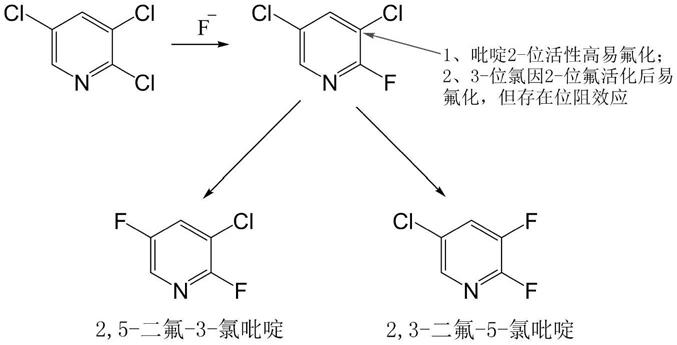
现有2,3-二氟-5-氯吡啶的合成方法中,均以2,3,5-三氯吡啶为原料,以氟化铯、氟化钾、氟化钠等为氟化试剂,以环丁砜、dmso、dmi等溶剂为介质,采用四丁基溴化铵、四苯基溴化膦、苄基三乙基氯化铵、18-冠-6等催化剂在220~230℃进行氟化反应,但其反应选择性较低,收率低为60~80%,并生成大量2,5-二氟-3-氯吡啶杂质,该杂质沸点为136~137℃,与产品2,3-二氟-5-氯吡啶沸点135℃仅差1~2℃,通过精馏难以分离纯化,市售2,3-二氟-5-氯吡啶产品含量一般为96%左右,单杂含量高,含2,5-二氟-3-氯吡啶3%以上,进一步造成下游炔草酯产品生产中生成大量杂质,产品含量低还需重结晶纯化。
[0008]
因此现有2,3-二氟-5-氯吡啶的工艺普遍存在着选择性差、使用不便、生产成本高等缺陷。
技术实现要素:
[0009]
本发明的目的在于提供一种单杂含量低的2,3-二氟-5-氯吡啶的绿色生产工艺,解决其制备工艺中存在的单杂含量高、选择性差、三废量大、生产成本高等问题,采用双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐催化剂进行氟化反应制得,将异构体单一杂质
2,5-二氟-3-氯吡啶的含量控制在0.9-1.1%,目标产物含量在98%以上,该工艺方案绿色环保,产品含量高、收率高于90%。
[0010]
为了达到上述目的,本发明提供如下技术方案:
[0011]
一种单杂含量低的2,3-二氟-5-氯吡啶的绿色生产工艺,包括以下步骤:
[0012]
1)向带精馏装置的反应釜中加入极性非质子性溶剂、无水氟化钾和脱水溶剂,共沸回流分水;
[0013]
其中,所述极性非质子溶剂是1,3-二甲基咪唑啉酮、环丁砜和n-甲基吡咯烷酮中的至少一种;所述脱水溶剂是指沸点低于130℃、且不与水混溶的芳香烃类有机溶剂;
[0014]
2)回流分水结束后,蒸馏回收脱水溶剂,蒸净脱水溶剂后向釜内加入催化剂双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐,温度保持在180-200℃,然后向反应釜内连续打入熔化的2,3,5-三氯吡啶物料进行反应;
[0015]
随着2,3,5-三氯吡啶物料的打入,精馏塔顶开始精馏出产物,收集120~138℃馏分,该馏分中2,3-二氟-5-氯吡啶的含量96.5~97%,2,5-二氟-3-氯吡啶杂质含量0.9~1.1%,2,3,5-三氟吡啶含量1.9~2.1%;
[0016]
2,3,5-三氯吡啶进料结束后继续精馏至塔顶无馏分时反应结束,获得2,3-二氟-5-氯吡啶粗产品;
[0017]
3)反应结束后,将2,3-二氟-5-氯吡啶粗产品进行负压精馏纯化,真空度为0.09~0.095mpa,收集的40~45℃前馏分为2,3,5-三氟吡啶,含量98.0~98.5%;45~60℃的过渡馏分套用至下一批负压精馏纯化;收集60~70℃馏分为2,3-二氟-5-氯吡啶纯品,其中,2,3-二氟-5-氯吡啶含量98%以上,单一杂质2,5-二氟-3-氯吡啶含量0.9~1.1%。
[0018]
进一步,步骤3)反应结束后还包括步骤4):真空度为0.09~0.095mpa,蒸出大部分溶剂,将剩余物料降温后加入步骤1)或步骤2)所述脱水溶剂打浆、洗涤、过滤,滤液合并蒸出溶剂回用至下一批反应;过滤废盐滤饼耙干,再加热水溶解、活性炭脱色、过滤,滤液浓缩结晶、40~50℃过滤,滤饼干燥得含量在98%以上的氯化钾,滤液直接进行喷雾干燥回收氟化钾套用至下一批反应。
[0019]
又,步骤4)中,反应结束打浆过滤滤液,收集含有催化剂双-(n-1,3-二甲基-2-咪唑啉基)-氯化铵盐的滤液,回收利用;将滤饼耙干,再加水热溶、脱色、过滤,浓缩结晶、过滤、干燥得氯化钾副产。
[0020]
进一步,步骤2)中,蒸馏回收的脱水溶剂在下一批回流分水步骤中套用。
[0021]
优选地,步骤1)所述脱水溶剂中的芳香烃类非质子性有机溶剂为苯、甲苯、二甲苯或氯苯。
[0022]
又,所述反应底物2,3,5-三氯吡啶、氟化钾和催化剂的摩尔比为1:2.0~2.5:0.01~0.1。
[0023]
进一步,所述反应底物2,3,5-三氯吡啶、反应溶剂和脱水溶剂的重量比为1:1~3:1.0~1.5。
[0024]
发明人研究发现,常规的2,3,5-三氯吡啶氟化反应体系多采用四丁基溴化铵、四苯基溴化膦、苄基三乙基氯化铵、18-冠-6等催化剂,催化活性较低,3-位氟交换反应时位阻效应较明显,造成生成物中2,5-二氟-3-氯吡啶含量较高,反应机理如下:
[0025][0026]
本发明中,以2,3,5-三氯吡啶为原料,氟化钾为催化剂,采用双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐催化剂进行氟化反应,反应式如下:
[0027][0028]
其中,cnc催化剂为自制双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐催化剂,由于该催化剂基团两端咪唑环的作用,其活性中心位阻小,氮正离子容易与氟离子结合转运,从而具有良好的活性,在本发明的氟化反应中,cnc可使2,3,5-三氯吡啶中2-位氟原子带来的位阻效应变小,3-位上氟交换反应时位阻效应减弱,从而减少因位阻效应氟化产生的异构体2,5-二氟-3-氯吡啶杂质,使该单一杂质的含量显著降低。
[0029]
另有部分底物生成全氟化产物2,3,5-三氟吡啶,但其沸点低,易于分离除去,且可作为医药中间体外售,反应接收的粗产品经二次精馏,可制得98%以上含量2,3-二氟-5-氯吡啶产品。
[0030]
本发明中对溶剂回收套用,废盐回收处理为氯化钾副产外售和氟化钾回用反应,反应时副产2,3,5-三氟吡啶可用于医药中间体外售,保证了所有物料的资源化利用,减少三废排放,降低了生产成本,工艺绿色环保。
[0031]
与现有技术相比,本发明的有益效果为:
[0032]
1)本发明在2,3,5-三氯吡啶的氟化反应体系中加入了新型相转移催化剂双-(n-1,3-二甲基-2-咪唑啉基)-氯化铵盐进行氟化反应,提高反应选择性,降低反应温度,同时配合一边进料一边精馏出产品的方案,显著减少物料在反应体系中停留时间,大大减少焦化、聚合等副反应,将反应收率提高至90%以上,粗产品经二次精馏制得98%以上含量2,3-二氟-5-氯吡啶产品,可显著降低下游炔草酯产品生成中产生的杂质,单一异构体杂质2,5-二氟-3-氯吡啶的含量降低了60%以上,有效提高产品含量和收率,降低成本。
[0033]
2)本发明在反应后处理中,直接采用回流分水溶剂打浆回收的含有催化剂双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐的滤液,可以直接套用至下一批反应,套用溶剂后只需补加少量催化剂就可以保证反应正常进行,降低生产单耗和成本。
[0034]
3)本发明将滤饼耙干的过程中,可以回收反应溶剂,降低生产成本。废盐滤饼耙干后,再加热水溶解、活性炭脱色、过滤,滤液浓缩结晶、过滤,滤饼干燥得含量在98%以上的氯化钾,滤液直接进行喷雾干燥回收氟化钾套用下一批反应。本发明使用的溶剂均回收套用,产生的废盐最终转化为氯化钾副产外售,使所有物料资源化利用,不产生废盐固废,绿
色环保。
附图说明
[0035]
图1为本发明实施例1中的产品含量gc分析图谱。
[0036]
图2为对比例1中的纯品含量gc分析图谱。
具体实施方式
[0037]
以下结合具体实施例对本发明作进一步说明。
[0038]
实施例1
[0039]
向干燥带精馏柱的反应釜中投入1,3-二甲基-2-咪唑啉酮(dmi)350kg和无水氟化钾(230kg,3.92kmol),再加入甲苯200kg回流分水约3小时,待分水结束,将甲苯蒸出回收,套用至下一批次反应。
[0040]
向上述反应体系中投入双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐(20kg,0.08kmol),升温至180~185℃,经泵向反应釜连续打入2,3,5-三氯吡啶(300kg,1.61kmol)熔化物料,随着原料打入,精馏塔顶开始精馏出产物,收集120~138℃馏分,当2,3,5-三氯吡啶物料进料结束后继续精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品229kg。接收的粗产品进行二次负压精馏纯化,真空度0.09~0.095mpa,控制塔顶采出回流比1:3,收集40~45℃前馏分,得2,3,5-三氟吡啶4.2kg,含量98.5%,塔顶升温至45~60℃接收过渡馏分套用下一批负压精馏纯化,塔顶升温至60~70℃收集馏分为2,3-二氟-5-氯吡啶纯品223kg(1.47kmol),产品含量98.7%,2,5-二氟-3-氯吡啶杂质含量1.0%,摩尔收率91.4%,产品含量分析图谱参见图1及表1。
[0041]
表1
[0042]
峰号保留时间高度高度%面积面积%12.18437930.036297100.04122.95818970.01836230.00533.16968490.065594190.08243.42327390.02657970.00853.58529500.02865220.00964.43912640.01243480.00675.302982530293.2537151811298.69785.4156587226.2527282461.00595.56279020.075202890.028106.256248650.236862300.119总计 1053617810072462296100
[0043]
反应结束后反应釜中真空度0.09~0.095mpa蒸出溶剂250kg,剩余物料降温后加入甲苯200kg溶剂打浆、洗涤、过滤,滤液合并蒸出溶剂回用至下一批反应中的分水步骤,含反应溶剂dmi和甲苯;过滤废盐滤饼干燥后,再加350kg热水溶解、5kg活性炭脱色、过滤,滤液浓缩结晶、40~50℃过滤,滤饼干燥得白色氯化钾结晶产品217kg(含量98.1%),滤液直接进行喷雾干燥回收氟化钾套用至下一批反应。
[0044]
实施例2利用回收的溶剂和氟化钾投料制备2,3-二氟-5-氯吡啶
[0045]
向干燥带精馏柱的反应釜中投入实施例1中回收的1,3-二甲基-2-咪唑啉酮(dmi)300kg和甲苯200kg,实施例1中回收的无水氟化钾(195kg,3.32kmol),回流分水约3小时,待分水结束,将甲苯蒸出回收,套用于下一次反应。
[0046]
向上述反应体系中投入双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐(5kg,0.01kmol),升温至190~195℃,经泵向反应釜连续打入2,3,5-三氯吡啶(300kg,1.61kmol)熔化物料,随着原料打入,精馏塔顶开始精馏出产物,收集120~138℃馏分,当2,3,5-三氯吡啶物料进料结束后继续精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品230kg。接收的粗产品进行二次负压精馏纯化,控制塔顶采出回流比1:3,收集40~45℃前馏分,得2,3,5-三氟吡啶4.6kg,含量98.3%,塔顶升温至45~60℃接收过渡馏分套用下一批负压精馏纯化,塔顶升温至60~70℃收集馏分为2,3-二氟-5-氯吡啶纯品221.6kg(1.46kmol),产品含量98.4%(2,5-二氟-3-氯吡啶杂质含量1.01%),摩尔收率90.5%。
[0047]
反应结束后反应釜中真空度0.09~0.095mpa蒸出溶剂220kg,剩余物料降温后加入甲苯200kg溶剂打浆、洗涤、过滤,滤液合并蒸出溶剂回用下一批反应;过滤废盐滤饼干燥后,再加300kg热水溶解、2kg活性炭脱色、过滤,滤液浓缩结晶、40~50℃过滤,滤饼干燥得白色氯化钾结晶产品215kg(含量98%),滤液直接进行喷雾干燥回收氟化钾套用至下一批反应。
[0048]
实施例3
[0049]
向干燥带精馏柱的反应釜中投入环丁砜300kg和无水氟化钾(200kg,3.41kmol),再加入甲苯250kg回流分水约3小时,待分水结束,将甲苯蒸出回收,套用于下一次反应。
[0050]
向上述反应体系中投入双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐(8kg,0.02kmol),升温至195~200℃,经泵向反应釜连续打入2,3,5-三氯吡啶(300kg,1.61kmol)熔化物料,随着原料打入,精馏塔顶开始精馏出产物,收集120~138℃馏分,当2,3,5-三氯吡啶物料进料结束后继续精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品226kg。接收的粗产品进行二次负压精馏纯化,控制塔顶采出回流比1:3收集40~45℃前馏分,得2,3,5-三氟吡啶4.5kg,含量98.6%;塔顶升温至45~60℃接收过渡馏分套用下一批负压精馏纯化,塔顶升温至60~70℃收集馏分为2,3-二氟-5-氯吡啶纯品223.6kg(1.47kmol),产品含量98.05%(2,5-二氟-3-氯吡啶杂质含量约1.05%),摩尔收率91%。
[0051]
反应结束后反应釜真空度0.09~0.095mpa蒸出溶剂200kg,剩余物料降温后加入甲苯250kg溶剂打浆、洗涤、过滤,滤液合并蒸出溶剂回用下一批反应;过滤废盐滤饼干燥后,再加300kg热水溶解、2kg活性炭脱色、过滤,滤液浓缩结晶、40~50℃过滤,滤饼干燥得白色氯化钾结晶产品210kg(含量98%),滤液直接进行喷雾干燥回收氟化钾套用至下一批反应。
[0052]
实施例4
[0053]
向干燥带精馏柱的反应釜中投入n-甲基吡咯烷酮300kg和无水氟化钾(200kg,3.41kmol),再加入二甲苯200kg回流分水约2小时,待分水结束,将甲苯蒸出回收,套用于下一次反应。
[0054]
向上述反应体系中投入双-(n,n
’‑
1,3-二甲基-2-咪唑啉基)-氯化铵盐(10kg,0.025kmol),升温至195~200℃,经泵向反应釜连续打入2,3,5-三氯吡啶(300kg,
1.61kmol)熔化物料,随着原料打入,精馏塔顶开始精馏出产物,收集120~138℃馏分,当2,3,5-三氯吡啶物料进料结束后继续精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品230kg。接收的粗产品进行二次负压精馏纯化,控制塔顶采出回流比1:3收集40~45℃前馏分,得2,3,5-三氟吡啶4.1kg,含量98.7%;塔顶升温至45~60℃接收过渡馏分套用下一批负压精馏纯化,塔顶升温至60~70℃收集馏分为2,3-二氟-5-氯吡啶纯品222kg(1.46kmol),产品含量98.45%(2,5-二氟-3-氯吡啶杂质含量约0.99%),摩尔收率90.7%。
[0055]
反应结束后反应釜真空度0.09~0.095mpa蒸出溶剂200kg,剩余物料降温后加入二甲苯200kg溶剂打浆、洗涤、过滤,滤液合并蒸出溶剂回用下一批反应;过滤废盐滤饼干燥后,再加400kg热水溶解、3kg活性炭脱色、过滤,滤液浓缩结晶、40~50℃过滤,滤饼干燥得白色氯化钾结晶产品208kg(含量98%),滤液直接进行喷雾干燥回收氟化钾套用至下一批反应。
[0056]
对比例1
[0057]
向干燥带精馏柱的反应釜中投入环丁砜300g和无水氟化钾(200g,3.41mol),再加入甲苯200g回流分水约3小时,待分水结束,将甲苯蒸出回收,套用于下一次反应。
[0058]
向上述反应体系中投入2,3,5-三氯吡啶(300g,1.61mol)和四苯基溴化膦(30g,0.07mol),需升温至210~220℃,精馏塔顶开始精馏出产物,收集120~140℃馏分,精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品195g。
[0059]
接收的粗产品采用与本发明相同的工艺进行二次负压精馏纯化,控制塔顶采出回流比1:3收集40~45℃前馏分,得少量2,3,5-三氟吡啶,塔顶升温至45~60℃接收过渡馏分套用下一批负压精馏纯化,塔顶升温至60~70℃收集馏分,获得2,3-二氟-5-氯吡啶纯品184g(1.2mol),产品含量96.2%,其中,2,5-二氟-3-氯吡啶杂质含量3.1%,摩尔收率75%,纯品含量分析图谱参见图2及表2。
[0060]
表2
[0061]
峰号保留时间高度高度%面积面积%12.957298290.203488030.04823.161243890.166496720.04933.4338020.00520350.00244.04231820.158477800.04755.3211309096589.1839736364296.68565.439148824210.13931354603.11475.58298370.067254280.02585.8594630.00311830.00196.0368410.00622840.002106.279101770.069258850.026总计 14678728100100702173100
[0062]
对比例2
[0063]
向干燥带精馏柱的反应釜中投入环丁砜300g和无水氟化钾(200g,3.41mol),再加入甲苯200g回流分水约3小时,待分水结束,将甲苯蒸出回收,套用于下一次反应。
[0064]
向上述反应体系中投入四苯基溴化膦(30g,0.07mol),升温至195~210℃,经泵向
反应釜连续打入2,3,5-三氯吡啶(300g,1.61mol)熔化物料,随着原料打入精馏塔顶开始精馏出产物,开始时出料量很少,反应后期收集120~140℃馏分,当2,3,5-三氯吡啶物料进料结束后继续精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品180g。
[0065]
接收的粗产品进行二次负压精馏纯化,控制塔顶采出回流比1:3收集40~45℃前馏分,得少量2,3,5-三氟吡啶,塔顶升温至45~60℃接收过渡馏分,套用下一批负压精馏纯化,塔顶升温至60~70℃时收集馏分,获得2,3-二氟-5-氯吡啶纯品175g(1.13mol),产品含量96.3%,其中,2,5-二氟-3-氯吡啶杂质含量3.0%,摩尔收率70%。
[0066]
对比例3
[0067]
向干燥带精馏柱的反应釜中投入1,3-二甲基-2-咪唑啉酮(dmi)300g和无水氟化钾(220g,3.75mol),再加入甲苯300g回流分水约3小时,待分水结束,将甲苯蒸出回收,套用于下一次反应。
[0068]
向上述反应体系中投入2,3,5-三氯吡啶(300g,1.61mol)和四丁基溴化铵(30g,0.092mol),需升温至200~210℃,精馏塔顶开始精馏出产物,收集120~140℃馏分,精馏至塔顶无馏分时反应结束,接收2,3-二氟-5-氯吡啶粗产品170g。
[0069]
接收的粗产品进行二次负压精馏纯化,控制塔顶采出回流比1:3收集40~45℃前馏分,得少量2,3,5-三氟吡啶,塔顶升温至45~60℃接收过渡馏分套用下一批负压精馏纯化,塔顶升温至60~70℃收集馏分为2,3-二氟-5-氯吡啶纯品164.8g(1.06mol),产品含量96.5%,其中,2,5-二氟-3-氯吡啶杂质含量2.9%,摩尔收率66%。