1.本发明涉及光致变色材料领域,具体涉及一种多取代双苯并色烯类化合物的制备方法。
背景技术:
2.光致变色是指当向某些化合物照射包含紫外线的光时,颜色迅速改变,当停止照射光而置于暗处时,就恢复到原来的颜色的可逆作用的现象。具有该性质的化合物被称为光致变色化合物,光致变色材料在变色眼镜、光信息存储、分子开关以及防卫识别技术等领域具有广泛应用前景,一直是化学及材料科学领域的研究热点之一。#
3.已知报道萘并吡喃是在多或单色光如uv光影响下能够改变颜色的光致变色的化合物。照射停止时,或者在温度和/或不同于初始光的多或单色光的影响下,该化合物回到其初始颜色。萘并吡喃在各种领域均有应用,例如在眼镜片、隐形镜片、太阳镜、滤光片、光学照相机或其它光学器件以及观测装置、玻璃窗和装饰性物体的制造中。2h-色烯在某些情况下在uv照射后具有中性灰色或褐色,这在变色镜中使用时尤其感兴趣,因为它不要求使用不同颜色的染料混合物以获得所需色调。事实上,不同颜色的染料可能具有不同的抗uv老化特性,不同的褪色动力学或不同的热依存性,导致使用期间镜片的色调发生改变。例如,对于眼镜片,出于视觉舒适性和安全原因(例如开车时),非常需要光致变色制品在不存在uv光时快速脱色。
技术实现要素:
4.本发明研究者在研究萘并吡喃类化合物时,发现当在2h萘并[1,2-b]吡喃(苯并色烯)的苯环上引入吸电子基团时,化合物具有较短的褪色半衰期。并且当两个苯并色烯类化合物通过给电子基团o连结起来后,能显著缩短褪色半衰期并能提升耐老化性能。由此合成多种多取代双苯并色烯类化合物,并提供其制备方法。
[0005]
本发明的技术方案如下:
[0006]
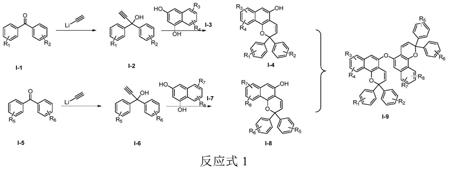
一种多取代双苯并色烯类化合物的制备方法,该化合物结构如下式所示:
[0007][0008]
其中:
[0009]
r1、r2、r5、r6各自选自氢、甲基、甲氧基、甲硫基、芳基、卤素、cn、no2、cf3或cf2h;
[0010]
r3、r4、r7、r8各自选自氢、甲基、甲氧基、甲硫基、卤素、cn、no2、cf3或cf2h,且r3、r4、r7、r8中至少一个为吸电子基团;
[0011]
该制备方法的反应式如下:
[0012][0013]
制备方法包括如下步骤:
[0014]
(1)将化合物i-1与乙炔基锂反应得到化合物i-2;
[0015]
(2)化合物i-2与化物i-3在樟脑酸存在下环合得到化合物i-4;
[0016]
(3)将化合物i-5与乙炔基锂反应得到化合物i-6;
[0017]
(4)化合物i-6与化物i-7在樟脑酸存在下环合得到化合物i-8;
[0018]
(5)将化合物i-4和i-8通过dead和三苯基膦条件下室温反应得到化合物i-9。
[0019]
如上所述的制备方法,优选地,所述化合物中r3、r7各自为h,r4、r8各自为f。
[0020]
如上所述的制备方法,优选地,所述化合物中r3、r7各自为h,r4、r8各自为cf3。
[0021]
如上所述的制备方法,优选地,所述化合物中r3、r7、r4、r8各自为f。
[0022]
如上所述的制备方法,优选地,所述步骤(1)的具体操作如下:将化合物i-1溶解于乙二胺中,加入乙炔锂乙二胺络合物,化合物i-1与乙炔锂乙二胺络合物的摩尔比为1∶(2~4);氮气气氛下,室温搅拌2~4小时;反应液用乙酸乙酯萃取,有机层用水、饱和氯化钠洗涤,干燥,浓缩,粗产物用硅胶柱色谱纯化,得到化合物i-2。
[0023]
如上所述的制备方法,优选地,所述步骤(2)的具体操作如下:将化合物i-2溶于甲苯,加入化合物i-3和樟脑磺酸,化合物i-2、化合物i-3和樟脑磺酸三者的摩尔比为1∶(1~1.5)∶(0.2~0.4);然后50~80℃搅拌2~4小时,反应结束后,浓缩,粗产物用硅胶柱色谱纯化,得到化合物i-4。
[0024]
如上所述的制备方法,优选地,所述步骤(3)的具体操作如下:将化合物i-5溶解于乙二胺中,加入乙炔锂乙二胺络合物,化合物i-5与乙炔锂乙二胺络合物的摩尔比为1∶(2~4);氮气气氛下,室温搅拌2~4小时;反应液用乙酸乙酯萃取,有机层用水、饱和氯化钠洗涤,干燥,浓缩,粗产物用硅胶柱色谱纯化,得到化合物i-6。
[0025]
如上所述的制备方法,优选地,所述步骤(4)的具体操作如下:将化合物i-6溶于甲苯,加入化合物i-7和樟脑磺酸,化合物i-6、化合物i-7和樟脑磺酸三者的摩尔比为1∶(1~1.5)∶(0.2~0.4);然后50~80℃搅拌2~4小时,反应结束后,浓缩,粗产物用硅胶柱色谱纯化,得到化合物i-8。
[0026]
如上所述的制备方法,优选地,所述步骤(5)的具体操作如下:将化合物i-4和i-8
溶于四氢呋喃中,加入偶氮二甲酸二乙酯和三苯基膦,化合物i-4、化合物i-8、偶氮二甲酸二乙酯和三苯基膦四者的摩尔比为1∶1∶(0.6~1.2)∶(0.6~1.2);反应12-24小时,反应结束后,浓缩,经硅胶制备板分离得到化合物i-9。
[0027]
本发明的反应式1中化合物i-4和化合物i-8可以为相同化合物,此时的制备反应如反应式2所示,包括如下步骤:
[0028]
(1)将化合物i-1与乙炔基锂反应得到化合物i-2;
[0029]
(2)化合物i-2与化和物i-3在樟脑酸存在下环合得到化合物i-4;
[0030]
(3)将化合物i-4溶于极性溶剂中,加入dead和三苯基膦,反应10-14小时,获得化合物i-10。
[0031][0032]
本发明中所用术语“烷基”是指具有1至8个碳原子的直链或支链的单价饱和烃基,其实例包括,但不限于甲基、乙基、1-丙基、2-丙基、1-丁基、2-甲基-1-丙基、2-丁基、2-甲基-2-丙基、叔丁基、1-己基、2-乙基丁基等。
[0033]
本发明中所用术语“环状烷基”是指3至8个碳原子的环烷基,但不限于环丙基、环丁基、环戊基、环己基、烷基取代环烷基。
[0034]
本发明中所用术语“芳基”其本身或作为另一取代基的一部分是指单价芳族烃基团,其由从母体芳族环体系的单个碳原子去掉一个氢原子而获得。芳基涵盖5-和6-元的碳环芳族环,例如,苯;双环环体系,其中至少一个环是碳环和芳族的,例如,萘、茚满和四氢萘;以及三环环体系,其中至少一个环是碳环和芳族的,例如,芴。芳基涵盖具有至少一个稠合到至少一个碳环芳族环、环烷基环、或者杂环烷基环的碳环芳族环的多环体系。
[0035]
本发明中所用术语“卤素”指氟、氯或溴。
[0036]
表示取代基从该处连接。
[0037]
本发明的有益效果在于:本发明的制备方法简单,产率较高。制备的通式(i-9)化合物具有显色灵敏度高、优良的耐久性和极短的褪色半衰期。具有多种变色材料的用途,例如,记忆材料、调光材料、光致变色透镜材料、光学过滤器材料、显示器材料、光信息器件、光开关元件、光刻胶材料、光量计或装饰材料等。
具体实施方式
[0038]
下列实施例用于说明而非限定通式(i-9)化合物的合成方法。温度均为摄氏度。如果没有另外说明,所有的蒸发均在减压下进行。如果没有另外说明,否则试剂是自商业供货商购得且未经进一步纯化即使用。终产物、中间体和原料的结构通过标准分析方法确认,例如元素分析、光谱特征分析,例如ms、nmr。使用的缩写是本领域常规缩写。
[0039]
中间体的制备:
[0040]
1.制备中间体a-5:10-氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇
[0041][0042]
(1)a-3:1,1-双(4-甲氧基苯基)丙-2-炔-1-醇的制备
[0043]
将4,4
’‑
二甲氧基二苯甲酮a-1(500mg,2.06mmol)溶解于10ml乙二胺中,加入乙炔锂乙二胺络合物a-2(558mg,6.20mmol)。氮气气氛下,室温搅拌2小时。反应结束后,用冰水淬灭,反应液用乙酸乙酯萃取。有机层用水、饱和氯化钠洗涤,无水硫酸钠干燥。浓缩,粗产物用硅胶柱色谱纯化,得到1,1-双(4-甲氧基苯基)丙-2-炔-1-醇a-3(450mg,白色固体),产率:81%。esi-ms m/z:269[m+h]
+
。
[0044]
(2)a-5:10-氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇的制备
[0045]
将1,1-双(4-甲氧基苯基)丙-2-炔-1-醇a-3(450mg,1.68mmol)溶于甲苯(10ml),加入8-氟萘-1,3-二醇a-4(358mg,2.01mmol)和樟脑磺酸(89mg,0.5mmol),然后60℃搅拌2小时,反应结束后,浓缩,粗产物用硅胶柱色谱纯化,得到10-氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇a-5(200mg,白色固体),产率:28%。esi-ms m/z:429[m+h]
+
。
[0046]
2.制备中间体a-7:10-三氟甲基-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇
[0047][0048]
将1,1-双(4-甲氧基苯基)丙-2-炔-1-醇a-3(450mg,1.68mmol)溶于甲苯(10ml),加入8-三氟甲基萘-1,3-二醇a-6(458mg,2.01mmol)和樟脑磺酸(89mg,0.5mmol),然后60℃搅拌2小时,反应结束后,浓缩,粗产物用硅胶柱色谱纯化,得到10-三氟甲基-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇a-7(180mg,白色固体),产率:22%。esi-ms m/z:479[m+h]
+
。
[0049]
3.制备中间体a-9:8,10-二氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇
[0050][0051]
将1,1-双(4-甲氧基苯基)丙-2-炔-1-醇a-3(450mg,1.68mmol)溶于甲苯(10ml),加入8,10-二氟萘-1,3-二醇a-8(349mg,2.01mmol)和樟脑磺酸(89mg,0.5mmol),然后60℃
搅拌2小时,反应结束后,浓缩,粗产物用硅胶柱色谱纯化,得到8,10-二氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇a-9(220mg,白色固体),产率:29%。esi-ms m/z:447[m+h]
+
。
[0052]
4.制备中间体a-12:10-氟-2,2-双(4-硫代甲基苯基)-2h-苯并[h]色烯-5-醇
[0053][0054]
将4,4
’‑
二甲硫基二苯甲酮a-10(500mg,1.82mmol)溶解于10ml乙二胺中,加入乙炔锂乙二胺络合物a-2(491mg,5.46mmol)。氮气气氛下,室温搅拌2小时。反应结束后,用冰水淬灭,反应液用乙酸乙酯萃取。有机层用水、饱和氯化钠洗涤,无水硫酸钠干燥。浓缩,粗产物用硅胶柱色谱纯化,得到1,1-双(4-甲硫基苯基)丙-2-炔-1-醇a-11(400mg,白色固体),产率:73%。esi-ms m/z:301[m+h]
+
。
[0055]
将1,1-双(4-甲硫基苯基)丙-2-炔-1-醇a-11(400mg,1.33mmol)溶于甲苯(10ml),加入8-氟萘-1,3-二醇a-4(284mg,1.6mmol)和樟脑磺酸(92mg,0.4mmol),然后60℃搅拌2小时,反应结束后,浓缩,粗产物用硅胶柱色谱纯化,得到10-氟-2,2-双(4-甲硫基苯基)-2h-苯并[h]色烯-5-醇a-12(174mg,白色固体),产率:28%。esi-ms m/z:461[m+h]
+
。
[0056]
实施例1:制备5,5
′‑
氧双(10-氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯)
[0057][0058]
将10-氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇a-5(200mg,0.47mmol)溶于四氢呋喃(5ml)中,加入偶氮二甲酸二乙酯(dead,35mg,0.2mmol)和三苯基膦(52mg,0.2mmol),反应16小时,反应结束后,浓缩,经硅胶制备板分离得到5,5
′‑
氧双(10-氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯)(75mg,白色固体),产率:38%。esi-ms m/z:839[m+h]
+
。
[0059]1h-nmr(400mhz,dmso-d6):δ7.83-7.62(m,2h),7.47-7.32(m,10h),7.30-7.15(m,2h),6.88-6.70(m,10h),6.58(d,j=7.2hz,2h),6.39(d,j=7.2hz,2h),3.52(s,12h)。
[0060]
实施例2:制备5,5
′‑
氧双(10-三氟甲基-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯)
[0061][0062]
将10-三氟甲基-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇a-7(200mg,0.42mmol)溶于四氢呋喃(5ml)中,加入偶氮二甲酸二乙酯(dead,31mg,0.18mmol)和三苯基膦(47mg,0.18mmol),反应16小时,反应结束后,浓缩,经硅胶制备板分离得到5,5
′‑
氧双(10-三氟甲基-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯)(50mg,白色固体),产率:23%。esi-ms m/z:939[m+h]
+
。
[0063]1h-nmr(400mhz,dmso-d6):δ8.12-7.65(m,2h),7.55-7.37(m,10h),7.32-7.12(m,2h),6.89-6.72(m,10h),6.57(d,j=7.2hz,2h),6.41(d,j=7.2hz,2h),3.55(s,12h)。
[0064]
实施例3:制备5,5
′‑
氧双(8,10-二氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯)
[0065][0066]
将8,10-二氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯-5-醇a-9(200mg,0.45mmol)溶于四氢呋喃(5ml)中,加入偶氮二甲酸二乙酯(dead,39mg,0.22mmol)和三苯基膦(58mg,0.22mmol),将反应液室温16小时,反应结束后,浓缩,经硅胶制备板分离得到5,5
′‑
氧双(8,10-二氟-2,2-双(4-甲氧基苯基)-2h-苯并[h]色烯)(42mg,白色固体),产率:21%。esi-ms m/z:875[m+h]
+
。
[0067]1h-nmr(400mhz,dmso-d6):δ7.92-7.72(m,2h),7.45-7.31(m,8h),7.33-7.12(m,2h),6.91-6.73(m,10h),6.57(d,j=7.2hz,2h),6.38(d,j=7.2hz,2h),3.52(s,12h)。
[0068]
实施例4:制备5,5
′‑
氧双(10-氟-2,2-双(4-硫代甲基苯基)-2h-苯并[h]色烯)
[0069][0070]
将10-氟-2,2-双(4-甲硫基苯基)-2h-苯并[h]色烯-5-醇a-12(200mg,0.43mmol)溶于四氢呋喃(5ml)中,加入偶氮二甲酸二乙酯(dead,38mg,0.22mmol)和三苯基膦(57mg,0.22mmol),将反应液室温16小时,反应结束后,浓缩,经硅胶制备板分离得到5,5
′‑
氧双(10-氟-2,2-双(4-甲硫基苯基)-2h-苯并[h]色烯)(35mg,白色固体),产率:18%。esi-ms m/z:903[m+h]
+
。
[0071]1h-nmr(400mhz,dmso-d6):δ7.82-7.62(m,2h),7.47-7.32(m,10h),7.30-7.15(m,2h),6.88-6.70(m,10h),6.58(d,j=7.2hz,2h),6.38(d,j=7.2hz,2h),2.42(s,12h)。
[0072]
实施例5:制备光致变色材料
[0073]
分别取实施例1-4得到的色烯化合物0.04质量份、二甲基丙烯酸四甘醇酯13质量份、2,2-双[4-(甲基丙烯酰氧基乙氧基)苯基]丙烷48质量份、聚乙二醇单烯丙基醚2质量份、三羟甲基丙烷三甲基丙烯酸酯20质量份、甲基丙烯酸缩水甘油酯9质量份、α-甲基苯乙烯6质量份、a-甲基苯乙烯二聚物2质量份、以及作为聚合引发剂的叔丁基过氧化2-乙基己酸酯1质量份充分混合,制备光致变色固化性组合物。接着,将得到的光致变色固化性组合物注入到由玻璃板和乙烯-乙酸乙烯酯共聚物制成的垫片(gasket)构成的模具中,进行铸塑聚合。聚合过程如下:使用空气炉,用18小时缓慢地从30℃升温至90℃,在90℃下保持2小时。聚合结束后,将聚合物从模具的玻璃模子中取出,得到由实施例1-4化合物制备的光致变色材料样品。
[0074]
实验例1:溶液中光致变色特性评价
[0075]
采用in mass法进行光致变色固化物(光致变色光学制品)的评价。将实施例5得到的各组聚合物(厚2mm,光致变色固化物(光学制品))作为试样,将光照射时间设定为1秒,除此以外,评价光致变色特性和退色半衰期。其结果示于表1。
[0076]
最大吸收波长(amax):为由(株)大塚电子工业制的分光光度计(瞬间多通道光电探测器mcpd2000m)求得的显色后的最大吸收波长,是显色时的色调的指标。
[0077]
显色浓度(abs):为上述最大吸收波长中的光照射0.5秒后的吸光度,是显色浓度的指标。可以说,该值越高,由光照射引起的着色变化越大,光致变色性越好。
[0078]
退色半衰期(t1/2):为当停止照射光时,试样的上述最大吸收波长中的吸光度降低至一半的值所需要的时间,是退色速度的指标。该时间越短,表示褪色速度越快。
[0079]
黄变度(yi):为了评价聚合固化后的黄变度,对于上述聚合固化后的试样,使用试验机(株)制的色差计(sm-4)测定色差。yi的值越小,聚合固化体(包括固化膜)的透明度越高,或评价化合物的劣化度越小。
[0080]
残存率(a
50
/a0x 100):为了评价光照射引起的显色的耐久性,进行以下的劣化促
进试验。通过试验机(株)制氙弧耐候机x25使得到的聚合物(试样)进行促进劣化50小时。其后,在试验的前后进行上述显色浓度的评价,测定试验前的显色浓度(a0)及试验后的显色浓度(a
50
),将其比(a
50
/a0)作为残存率,设定为显色的耐久性的指标。残存率越高,显色的耐久性越高。
[0081]
表1:
[0082][0083]
对比例1
[0084]
为了进一步比较,采用实施例5相同的方法,用以下式a~式f化合物制备光致变色固化薄膜,用实验例1方法评价得到的光致变色塑料透镜的特性,其结果见表2:
[0085]
[0086][0087]
表2
[0088]
[0089][0090]
由实验结果表明,本发明化合物在引入吸电子基团的同时将不同取代的苯并色烯通过给电子基团o连接,发现该类化合物具有实用的退色半衰期,和良好的耐老化性能,具有一旦停止光照射颜色瞬间消失的光致变色性。
[0091]
因此,当使用本发明的多取代双苯并色烯类化合物制作光致变色材料,例如光致变色透镜时,可以制造出具有这样一种性质的光致变色透镜,当来到室外时迅速显色,从室外返回室内时迅速退色并恢复到原来的色调,并且可以长时间使用。
[0092]
本发明的多取代双苯并色烯类化合物由于显示出以上优异的效果,可适用于各种各样的用途,例如,调光材料、全息照相材料、油墨材料、光信息器件、光开关元件、以及光刻胶材料等。