1.本发明实施例涉及合成冠醚领域,具体涉及一种连续合成18-冠醚-6的反应工艺和装置。
背景技术:
2.冠醚是分子中含有多个-氧-亚甲基-结构单元的大环多醚,命名时把环上所含原子的总数标注在“冠”字之前,其中所含氧原子数标注在名称之后。常见的冠醚有12-冠-4、15-冠-5、18-冠-6,24-冠-8和30-冠-10等,这是杜邦公司的pedersen在1967年意外发现的,并发现这类化合物具有许多异常的特性,冠醚的空穴结构对离子有选择作用,在有机反应中可作催化剂。目前,已合成的冠醚有数百种,但实际被应用的却只有三十几种,其中产量最大的是18-冠-6(1,4,7,10,13,16-六氧环十八烷c
12h24
o6)。它为白色晶体,熔点38-39.5℃,沸点116℃/26.6pa。
3.冠醚最主要的特点之一,是可与各种金属盐、铵盐、有机阳离子化合物等形成稳定的络合物。利用这一性质,可将各种盐类溶于有机溶剂。冠醚可将阳离子螯环内,同时由于有朝外的有机基因可生成络合物,亦可溶于非极性的有机溶剂。这时,未被溶剂化的阴离子,以裸露的阴离子形式存在于溶剂中,故活性极大。冠醚可使碱金属和有机碱金属化合物溶解在有机溶剂中。因而在有机合成、光学拆分、重金属螯合、分离、分析以及生理活性的医药、生物化学等方面找到广泛的应用。作为在有机合成中最常被用作相转移催化剂的18-冠-6,其合成方法大致可以分为两类,一类是路易斯酸(bf3·
hf)作催化剂,在kbf4盐存在下使环氧乙烷进行齐聚反应,虽然原料简单但是操作特别麻烦而且副反应多,因此目前工业上普遍采用传统的合成通用醚的williamson反应(威廉森合成法)。
4.比较实用的威廉森合成法利用低聚甘醇和低聚甘醇的二卤化物或者二甲苯磺酸酯化合物在碱金属离子作为模板反应而成,主要有三种不同形式:(1)由低聚甘醇的二氯化物和低聚甘醇合成,如cn 103275059a中批露以三甘醇、二氯代三甘醇和氢氧化钾作为反应物制备而得,并且在反应后反复进行回流、蒸馏和重结晶过程。通过控制反应过程的时间、温度和不同反应物的摩尔比可以提高18-冠醚-6的产量,也使得杂质少、纯度高。cn111087382a所述制备方法,包括冠醚粗品的制备、冠醚粗品的提纯;所述粗品的制备,包括三甘醇、四氢呋喃、二氯三甘醇一次性加料、分批加入碱、保温反应,结合其他工艺过程提高反应收率,使用乙腈配合-解配的方法提纯产品,收率35%以上,纯度99.8%以上。但反应时间偏长(20h以内);粗品提纯结晶时间也偏长(小于10h)。(2)由低聚甘醇和对甲苯磺酰氯在碱性溶液中合成,cn110759886a该方法也采用传统间歇搅拌反应器,将对甲苯磺酰氯滴加至到盛有三甘醇和氢氧化钾的质子溶剂中,保证了较高的反应速率、反应物转化率的同时,也简化了后续分离操作。反应温度为30-80℃,反应时间为2-6h;然后加入分离剂除去副产物,控制反应温度为70-100℃,反应时间为3-6h;最后经后处理得到18-冠醚-6。(3)由低聚甘醇和低聚甘醇的二甲苯磺酸酯(tosylate)合成,如cn 108409706 a中批露在工业微波反应器中间歇操作对甲基苯磺酸二元醇酯、二元醇、碱的混合物,然后经除盐分离、萃取、蒸
除溶剂、减压蒸馏等后处理步骤得到粗产物18-冠醚-6。本工艺由于直接以甲基苯磺酸二元醇酯作为原料之一,因此可以降低反应温度,缩短反应时间(0℃~40℃,10分钟至5小时)。和方法(2)相比实际上合成步骤中多出合成和分离纯化甲基苯磺酸二元醇酯中间体的额外一步,换句话讲,该原料并不是简单易得。
5.前两种方法在实际生产中,反应产率低于60%,都采用普通的加热搪瓷反应釜,需要长时间的回流加热,也需要复杂的后处理操作,造成原料与人工成本高,能耗高、而产量有限。第三种方法虽然使用工业微波反应器可以大幅缩短反应时间降低反应温度,但是依旧是间歇批次操作方式,而且需要额外制备低聚醇的二甲苯磺酸酯中间体。因此,目前工业生产上急需要一种连续合成18-冠醚-6的反应工艺和装置,主要解决现有生产工艺间歇操作耗时长、选择性低或者多步间歇工艺需要中间体的额外分离纯化等问题。
6.随着微反应器技术在我国医药精细化工领域的推广和成功案例的指引,为了方便实际生产需要,特开发一种连续合成18-冠醚-6的反应工艺和装置,适用于工业化生产,能大幅降低成本、节能、并提高产量,有很显著地经济效益。
技术实现要素:
7.为此,本发明实施例提供一种连续合成18-冠醚-6的反应工艺和装置,主要解决现有合成工艺间歇操作能耗高而且产率不高或者有些工艺中间体不易得而需要额外合成的问题。通过本发明,可以采用连续微通道工艺替换现有的间歇搅拌方式,不仅可以大幅幅降低成本、节能、并提高产量,而且可以快速放大适合工业化生产。
8.为了实现上述目的,本发明实施例提供如下技术方案:一种连续合成18-冠醚-6的反应工艺,包括以下步骤:
9.步骤一:由二元醇i(ho(ch2ch2o)mh)和对甲苯磺酰氯或甲磺酰氯现场生成对应的甲基苯磺酸二元醇酯或者甲磺酸二元醇酯;
10.步骤二:该中间体不经过分离纯化直接和相应二元醇ii(ho(ch2ch2o)nh)在钾离子模板作用下环化反应合成目标产物,其中m和n均为大于或等于1的整数,范围为m+n≤6,优选m+n等于6。
11.进一步地,所述步骤一中的二元醇i选自乙二醇、聚乙二醇、二甘醇、三甘醇、四甘醇、五甘醇、聚甘醇之一,所述步骤二中的二元醇ii选自乙二醇、聚乙二醇、二甘醇、三甘醇、四甘醇、五甘醇和聚甘醇之一。
12.进一步地,所述步骤一中的二元醇i和对甲苯磺酰氯或者甲磺酰氯的进料实时摩尔比为1:2.0~2.4,所述步骤二中的二元醇ii和第一步中二元醇i的进料实时摩尔比为1:1.0~1.2。
13.进一步地,所述步骤一中的二元醇i和除对甲苯磺酰氯或甲磺酰氯以外的附酸剂、添加剂和/或溶剂组成第一预混合物,对甲苯磺酰氯或甲磺酰氯和溶剂组成第二预混合物;所述步骤二中的二元醇ii和所需要的碱、可能的催化剂和溶剂组成第三预混合物。
14.进一步地,所述步骤一种酯化反应工艺中使用的附酸剂选自三乙胺、n,n
’‑
二异丙基乙基胺、氢氧化钾、氢氧化钠、碳酸钾等之一,所述步骤二中环化工艺中采用的碱优选包含钾离子或者铵离子的模板试剂,包括氢氧化钾、叔丁醇钾、氢氧化钠、甲醇钠、碳酸钾、醋酸钾、蚁酸钾等。
15.进一步地,所述步骤一种酯化反应工艺中使用的附酸剂和二元醇i的进料实时摩尔比为1.0~1.2:1,所述步骤二中的碱和第二步中二元醇ii的进料实时摩尔比为1.0~1.2:1;所述附酸剂或者碱相应的物质的量,均以二元附酸剂或二元碱来计算。
16.进一步地,所述为了加速步骤二模板环化反应,可以于反应初始在二元醇ii中加入催化量的产物18-冠醚-6,催化剂18-c-6占用底物二醇的摩尔比为0.1-10%,所述步骤一中酯化反应工艺中使用的溶剂选自四氢呋喃、乙腈、二氧六环、甲苯之一或者不使用溶剂,所述步骤二中环化工艺中采用的溶剂选自二氧六环、甲苯、乙腈、四氢呋喃或水等。
17.进一步地,所述步骤一种工艺采用第一微混合器和第一微反应器串联,然后和步骤二工艺中第二微混合器其中一个进口相连,随后进入第二微反应器,最终得到目标产物,其中微混合器可以选自下面的一种或多种:t型、y型、套管式、梳式、层叠式、盘片式、环锥式、交叉指型式以及微孔涡流混合器、锥面盘片混合器、对撞流微混合器等,微反应器可以选自下面的一种或多种:毛细管式、层叠式、曲径式、夹心式和插片式微反应器,扁管涡流反应器、扁管插片反应器以及拜耳sandwich微反应器和康宁心形板式反应器等,为了防止微通道工艺堵塞以及工艺维护方便,其中的微混合器和微反应器优选可拆卸式,所述微混合器和微反应器的通道尺寸可以由亚微米到毫米级,优选10~500微米,其与传统的混合或反应装置相比,具有更大的比表面积/体积比,同时所述微混合器和微反应器均集成有用于换热的微换热器。
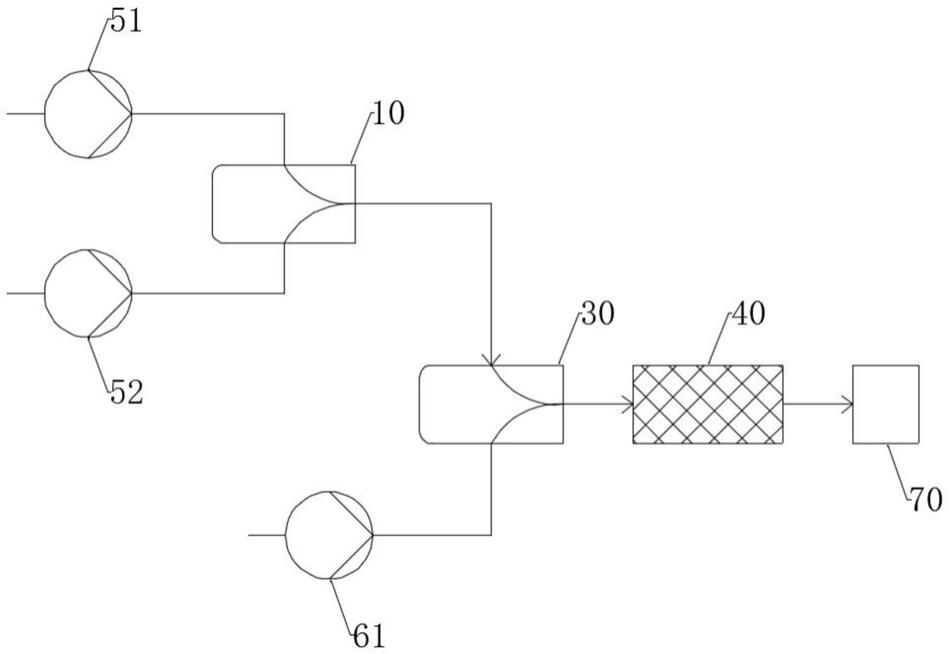
18.一种连续合成18-冠醚-6的反应装置,包括第一预混合物通过泵、第二预混合物通过泵、第一微混合器、第二微混合器、第二微反应器、通过泵与,所述第一预混合物通过泵与第二预混合物通过泵的输出端与第一微混合器连接,所述第一微混合器的输出端与第二微混合器连接,所述通过泵的输出端与第二微混合器连接,所述第二微混合器的输出端与第二微反应器连接,所述第二微反应器的输出端与连接。
19.一种连续合成18-冠醚-6的反应装置,包括第一预混合物通过泵、第二预混合物通过泵、第一微混合器、第一微反应器、第二微混合器、第二微反应器、通过泵与,所述第一预混合物通过泵与第二预混合物通过泵的输出端与第一微混合器连接,所述第一微混合器的输出端与第一微反应器连接,所述第一微反应器的输出端与第二微混合器连接,所述通过泵的输出端与第二微混合器连接,所述第二微混合器的输出端与第二微反应器连接,所述第二微反应器的输出端与连接。
20.一种连续合成18-冠醚-6的反应装置,包括第一加料罐、第二加料罐与混合器,所述第一加料罐与第二加料罐的输出端与混合器连接。
21.本发明实施例具有如下优点:
22.本发明所述的反应在连续流微通道反应器中进行,反应速度比常规方法要加快数十甚至数千倍,反应可以在室温下进行、反应时间超短(低于十分钟)、省人工、产率高、可采用水作溶剂,适用于工业化生产,能大幅降低成本、节能、并提高产量,有很显著地经济效益。本发明的原理是利用连续流微通道进行两步串联的williamson醚化反应。
附图说明
23.为了更清楚地说明本发明的实施方式或现有技术中的技术方案,下面将对实施方式或现有技术描述中所需要使用的附图作简单地介绍。显而易见地,下面描述中的附图仅
仅是示例性的,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图引伸获得其它的实施附图。
24.本说明书所绘示的结构、比例、大小等,均仅用以配合说明书所揭示的内容,以供熟悉此技术的人士了解与阅读,并非用以限定本发明可实施的限定条件,故不具技术上的实质意义,任何结构的修饰、比例关系的改变或大小的调整,在不影响本发明所能产生的功效及所能达成的目的下,均应仍落在本发明所揭示的技术内容得能涵盖的范围内。
25.图1是根据本发明的一个实施例的连续合成18-冠醚-6的反应工艺的装置示意图;
26.图2是根据本发明的另一个实施例的连续合成18-冠醚-6的反应工艺装置示意图。
27.图3是根据本发明的一个实施例的连续合成18-冠醚-6的反应工艺中制备第一、第二或第三预混合物的装置示意图。
28.图中:10、第一微混合器;20、第一微反应器;30、第二微混合器;40、第二微反应器;51、第一预混合物通过泵;52、第二预混合物通过泵;61、第三预混合物通过泵;100、第一加料罐;200、第二加料罐;300、混合器。
具体实施方式
29.以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
30.图1是根据本发明的一个实施例的连续合成18-冠醚-6的反应工艺的装置示意图。第一预混合物通过泵51与第二预混合物通过泵52后输入至第一微混合器10进行混合,然后与通过泵61的第三预混合物在第二微混合器30进行第二次混合。混合后的所述物料输入至第二微反应器40,用于延长反应停留时间并控制温度。反应结束后,将产物倒入产品收集器70中。
31.图2是根据本发明的另一个实施例的连续合成18-冠醚-6的反应工艺的装置示意图。与图1所述的合成18-冠醚-6的的装置的区别在于,在第一微混合器10和第二微混合器30之间串联有第一微反应器20,用于第一步工艺延长反应停留时间并控制温度。其他部分完全相同。
32.实施例1:
33.如图3所示,在室温下,将三甘醇、附酸剂三乙胺按摩尔比为1:2以及溶剂(混合后三甘醇底物的thf浓度为4摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第一预混合物;
34.此外在室温下,将对甲基苯磺酰氯(与底物三甘醇摩尔比为2:1)和溶剂thf混合为第二预混合物,浓度为8摩尔/升;
35.将三甘醇、碱氢氧化钾按摩尔比为1:2以及溶剂(混合后三甘醇底物的二氧六环浓度为4摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第三预混合物;
36.如图1所示,在室温下,将第一预混合物和第二预混合物分别经过泵51、泵52输入至微混合器10,第一预混合物和第二预混合物的体积流量分别为10.0ml/min和10.0ml/
min,总体积流量为20.0ml/min。该混合器10为交叉指型式,通道尺寸为85微米的微混合器。然后该混合物与经过泵61的第三预混合物在微混合器30中进行混合并发生反应,第三预混合物的体积流量为10.0ml/min;之后反应混合物经过微反应器40进行醚化反应。该混合器30为层叠式,通道尺寸为100微米的微混合器;反应器40为夹层式微反应器,其通道尺寸经内嵌的混合盘片分割为100微米;微混合器和微反应器集成的微换热器均无需通入换热介质。反应产物最后达到收集器70中。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率65%,总停留时间为8min。
37.实施例2:
38.与实施例1的区别在于:
39.如图2所示,在室温下,将第一预混合物和第二预混合物分别经过泵51、泵52输入至微混合器10,第一预混合物和第二预混合物的体积流量分别为10.0ml/min和10.0ml/min,总体积流量为20.0ml/min。该混合器10为交叉指型式,通道尺寸为85微米的微混合器。在进入第二微混合器30前,首先经过微反应器20进行充分的酯化反应现场生成对应的甲基苯磺酸二元醇酯,然后该混合物与经过泵61的第三预混合物在微混合器30中进行混合并发生后续连串醚化环化反应。各物料的进料体积流量保持一致,混合器与反应器的配置也不变。反应器20为扁管涡流式微反应器,反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率71%,总停留时间10min。
40.实施例3:
41.与实施例2的区别在于:
42.如图3所示,在室温下,将三甘醇、碱氢氧化钾按摩尔比为1:2以及溶剂(混合后三甘醇底物的二氧六环浓度为4摩尔/升),以及催化量的18-冠醚-6(和底物二醇的摩尔比1:100)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第三预混合物;
43.如图2所示,各物料的进料体积流量保持一致,混合器与反应器的配置也不变。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率76%,总停留时间7.5min。
44.实施例4:
45.与实施例3的区别在于:
46.如图2所示,两步工艺反应设定温度均为0℃,通过微混合器和微反应器集成的微换热器40中通入低温换热介质来控制实现。其他各物料的进料体积流量保持一致,混合器与反应器的配置也不变。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率69%,总停留时间10min。
47.实施例5:
48.与实施例3的区别在于:
49.如图2所示,两步工艺反应设定温度均为35℃,通过微混合器和微反应器集成的微换热器40中通入低温换热介质来控制实现。其他各物料的进料体积流量保持一致,混合器与反应器的配置也不变。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率78%,总停留时间6min。
50.实施例6:
51.与实施例3的区别在于:
52.如图2所示,微混合器和微反应器的配置不同。其中混合器10为锥面盘片式,通道尺寸为150微米的微混合器;微反应器20为扁管涡流式微反应器,通道尺寸最窄处为125微米;微混合器30为微孔涡流套管式,通道尺寸为100微米的微混合器;微反应器40为扁管插片式微反应器,其通道尺寸经内嵌的混合盘片分割为125微米;微混合器反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率73%,总停留时间8min。
53.实施例7:
54.如图3所示,在室温下,将二甘醇、附酸剂三乙胺按摩尔比为1:2以及溶剂(混合后二甘醇底物的thf浓度为8摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第一预混合物;
55.此外在室温下,将对甲基苯磺酰氯(与底物二甘醇摩尔比为2:1)和溶剂thf混合为第二预混合物,浓度为16摩尔/升;
56.将四甘醇、碱氢氧化钾按摩尔比为1:2以及溶剂(混合后四甘醇底物的二氧六环浓度为8摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第三预混合物;
57.如图1所示,在室温下,将第一预混合物和第二预混合物分别经过泵51、泵52输入至微混合器10,第一预混合物和第二预混合物的体积流量分别为5.0ml/min和5.0ml/min,总体积流量为10.0ml/min。该混合器10为对撞流式,通道尺寸为125微米的微混合器。之后经过微反应器20进行充分的酯化反应现场生成对应的甲基苯磺酸二元醇酯,然后该混合物与经过泵61的第三预混合物在微混合器30中进行混合并发生后续连串醚化环化反应。第三预混合物的体积流量为5.0ml/min;之后反应混合物经过微反应器40进行醚化反应。微反应器20为拜耳夹心式反应器;该混合器30为微孔涡流式,通道尺寸为100微米的微混合器;反应器40为康宁心形微反应器,其通道尺寸最窄处为100微米;微混合器和微反应器集成的微换热器均无需通入换热介质。反应产物最后达到收集器70中。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率68%,总停留时间12min。
58.实施例8:
59.与实施例7不同在于:
60.如图3所示,在室温下,将二甘醇、附酸剂n,n
‘‑
二甲基乙基胺按摩尔比为1:2以及溶剂(混合后二甘醇底物的thf浓度为8摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第一预混合物;
61.此外在室温下,将甲磺酰氯(与底物二甘醇摩尔比为2:1)和溶剂thf混合为第二预混合物,浓度为16摩尔/升;
62.将四甘醇、碱氢氧化钠按摩尔比为1:2以及溶剂(混合后四甘醇底物的二氧六环浓度为8摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第三预混合物;
63.其他各物料的进料体积流量保持一致,混合器与反应器的配置也不变。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率70%,总停留时间10min。
64.实施例9:
65.与实施例7不同在于:
66.如图3所示,在室温下,将乙二醇、附酸剂三乙胺按摩尔比为1:2.2以及溶剂(混合后乙二醇底物的甲苯浓度为12摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第一预混合物;
67.此外在室温下,将对甲苯磺酰氯(与底物乙二醇摩尔比为2.2:1)和溶剂甲苯混合为第二预混合物,浓度为24摩尔/升;
68.将五甘醇、碱氢氧化钾按摩尔比为1:2.2以及溶剂(混合后五甘醇底物的水溶液浓度为12摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第三预混合物;
69.其他各物料的进料体积流量保持一致,混合器与反应器的配置也不变。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率72%,总停留时间9min。
70.实施例10:
71.如图3所示,在室温下,将四甘醇、附酸剂氢氧化钾按摩尔比为1:2.4以及溶剂(混合后二醇底物的乙腈浓度为4摩尔/升)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第一预混合物;
72.此外在室温下,将对甲苯磺酰氯(与底物四甘醇摩尔比为2.4:1)和溶剂乙腈混合为第二预混合物,浓度为8摩尔/升;
73.将二甘醇、碱氢氧化钾按摩尔比为1:2.4以及溶剂(混合后二甘醇底物的乙腈溶液浓度为4摩尔/升),以及催化量的18-冠醚-6(和底物二醇的摩尔比3.5:100)分别从加料罐100和加料罐200加入到以电磁或机械搅拌的混合器300中混合,制得第三预混合物;
74.如图1所示,在室温下,将第一预混合物和第二预混合物分别经过泵51、泵52输入至微混合器10,第一预混合物和第二预混合物的体积流量分别为20.0ml/min和20.0ml/min,总体积流量为40.0ml/min。该混合器10为对撞流式,通道尺寸为180微米的微混合器。之后经过微反应器20进行充分的酯化反应现场生成对应的甲基苯磺酸二元醇酯,然后该混合物与经过泵61的第三预混合物在微混合器30中进行混合并发生后续连串醚化环化反应。第三预混合物的体积流量为20.0ml/min;之后反应混合物经过微反应器40进行醚化反应。微反应器20为拜耳夹心式反应器;该混合器30为微孔涡流式,通道尺寸为180微米的微混合器;反应器40为康宁心形微反应器,其通道尺寸最窄处为200微米;微混合器和微反应器集成的微换热器均无需通入换热介质。反应产物最后达到收集器70中。反应粗产物再经除盐分离、萃取、蒸除溶剂、减压蒸馏粗产物,得到18-冠醚-6,收率70%,总停留时间7min。
75.虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。