1.本发明属于微生物组或结直肠癌技术领域,具体涉及一种结直肠癌的微生物标志物及其应用。
背景技术:
2.结直肠癌(colorectal cancer,crc)作为全球第二大癌症相关死亡原因,每年约有90万死亡病例,并且其发病率在50岁以下人群中迅速上升。随着时间的推移,crc的经济负担持续增长,预计2018年仅美国而言,其经济负担就约为166.3亿美元。因此,改进结直肠癌的筛查方法尤为重要,这可能不仅受到遗传和表观遗传因素的影响,还受到环境因素的影响,其中包括已深入研究的肠道微生物组。
3.与健康人相比,crc患者的微生物群严重失调。多项研究已经揭示了crc和细菌组之间的密切联系,细菌组是肠道微生物组的主要组成部分。目前已知在结直肠癌患者的粪便含有大量的前致癌细菌,包括具核梭杆菌、大肠杆菌、脆弱拟杆菌和胃链球菌,同时大量的有益菌或保护菌属如梭状芽胞杆菌、玫瑰杆菌、粪杆菌和双歧杆菌则消失。
4.人体宿主被微生物群所定植,这些微生物群由多种多样的生物体组成,除了细菌之外,还包括真菌、古菌和病毒。此外,考虑到宿主生态位的共享,这些生物之间还存在有大量的物种间相互作用,这对人体健康或疾病状态产生了重大影响。非细菌物种是胃肠道稳定的共生体,参与多种代谢活动。据报道,古菌在甲烷生成、重金属转化和免疫调节中发挥关键作用。病毒,特别是噬菌体,显著影响细菌的生长和全球的生态平衡。真菌及其菌群,调节宿主的免疫系统,并且与细菌及其它微生物物种有密切的相互作用。所以,仅仅依靠单一物种的检测,无法对crc的诊断做出客观、精准和全面的系统性判断,必须结合多种物种,综合考虑各物种之间的相互作用关系。
5.a.目前临床常用的结直肠癌诊断方法主要包括x射线检查、肠镜检查和癌胚抗原(cea) 检查。这三类方法各有优缺点,其中x射线检查的可观察全部结直肠病变的影像,同时可了解肠道运动情况,协助少数疾病的治疗,但该方法受检查者的经验水平与设备条件影响较大,无法获取病理标本确诊,不能做治疗,并且对凹陷性及平坦性病变识别能力较差,有辐射损害。目前肠镜检查适应于筛查、确诊、随访与治疗,可谓全天候手段,其优点是可直接观察病变,范围可及全结肠,没有盲区,能同时取病理标本,做某些内镜下治疗,减少部分外科手术。该项检查被认为是结肠肿瘤诊断的重要手段。但该方法的缺点是术前准备,尤其是肠道清洁处理稍显麻烦。诊断准确性受内镜医师的经验与操作技术熟练程度影响甚大,创伤性也较大,有一定比例的并发症,主要是出血与穿孔,少数会发生心血管突发事件。无痛肠镜可减少不适感,但增加麻醉风险,且收费要高一些。癌胚抗原(cea)对早期病例的诊断价值不大;有研究表明,mir-92可以作为结直肠癌标志物,但是依据血液中mir-92的含量进行结直肠癌的诊断,错误率高达30%。随着人类基因组测序计划的完成以及高通量测序技术的发展,基因筛查技术已成为一种新的结直肠癌的诊断方法,在结直肠癌的早期诊断中具有显著的优势,其中宏基因组测序越来越受到普遍的欢迎。
6.b.已有的结直肠癌微生物标志物涉及的微生物种类少,且基本局限为单一物种如细菌,并未涉及四大界微生物之间的相互作用;
7.c.微生物组结构可能受基因、饮食、药物以及其他外界环境因素的影响,存在明显的个体间差异;
8.d.目前的微生物组研究大多集中在细菌,对于物种间的大规模相互作用并未触及,而且所得结果缺乏实验验证,因此很难全面代表疾病状态下的微生物组客观状态;
9.e.相较于传统的抗肿瘤治疗手段,微生物治疗已初步崭露头角,但特异性微生物种属应用于结直肠癌治疗的相关机制仍需要进一步研究;
10.f.目前结直肠癌微生物组学研究样本量均较小,所得结果不具有普适意义,需要大样本量和多中心合作以获得具有统计学意义的大样本数据;
11.g.目前的样本收集、dna提取和微生物组分析流程参差不齐,需要建立标准化的样本收集与处理流程,以减少因采样标准差异而造成的实验结果偏倚;
12.f.如何优化采样技术并避免临床样本的污染也是当前面临的问题之一。
13.未来,更多的研究成果将进一步阐明稀少和稀有微生物组尤其是真菌与结直肠癌的关系,这些机理将为结直肠癌发生和发展机制、微生物标志物早期筛查、精准和个体化治疗方案等方面提供新的理论基础和治疗靶标。
技术实现要素:
14.针对现有技术中的不足,迄今为止,只有少数研究探讨了非细菌肠道微生物的群落波动,以及疾病中多界微生物之间潜在的相互作用,如非酒精性脂肪性肝病、炎症性肠病。而且这些研究主要集中在单一界内物种的微生物相互作用上。crc相关微生物群的研究涉及多个界物种(细菌、真菌、古菌和病毒),特别是这些微生物之间的相互作用,目前仍处于起步阶段。
15.本发明所涉及的微生物囊括细菌、真菌、古菌和病毒,可以较为全面地综合各种微生物,对crc做出更为准确的判断。具体地,本发明涉及结直肠癌患者粪便样本中的细菌、真菌、古菌和病毒四大微生物类群标志物及不同物种间的特定相互作用关系,及其在结直肠癌筛查、诊断或辅助治疗中的应用。
16.为达到上述目的,本发明的解决方案是:
17.一种结直肠癌的微生物标志物,其选自细菌、真菌、古菌和病毒中的一种及以上。
18.优选地,结直肠癌的微生物标志物为细菌和真菌的组合。
19.一种上述的微生物标志物在制备结直肠癌诊断试剂和/或结直肠癌诊断药物中的应用。
20.一种药物组合物包括上述结直肠癌的微生物标志物。
21.一种诊断上述的微生物标志物的试剂盒,该试剂盒包括结直肠癌的微生物标志物。
22.一种预测结直肠癌的模型,其包括结直肠癌微生物标志物的丰度和微生物标志物间的相互作用。
23.优选地,结直肠癌微生物标志物的丰度测定方法为宏基因组测序。
24.人体宿主被微生物群所定植,这些微生物群由多种多样的生物体组成,除了细菌
之外,还包括真菌、古菌和病毒。考虑到宿主生态位的共享,这些生物之间的相互作用可以对人类健康或疾病状态产生重大影响。非细菌物种是胃肠道稳定的共生体,参与多种代谢活动。据报道,古菌在甲烷生成、重金属转化和免疫调节中发挥关键作用。病毒,特别是噬菌体,显著影响细菌的健康和全球生态循环。真菌及其菌群,调节宿主的免疫系统,并且与细菌有密切的相互作用。所以,仅仅依靠单一物种的检测,无法对crc的诊断做出精准的判断,必须结合多种物种,综合考虑各物种之间的相互作用关系。
25.本发明涉及提取结直肠癌患者粪便样本中的菌群dna通过宏基因组测序对物种进行鉴定,获得肠道微生物组(微生物组是细菌、真菌、古菌和病毒互作所形成的动态平衡体系)中四大微生物类群的种类和丰度特征,并通过微生物组成分析获得结直肠癌患者肠道微生物组中特异性富集的物种种类、数量、比值和相互作用特征,最终以此为基础进行结直肠癌的诊断。
26.由于采用上述方案,本发明的有益效果是:
27.第一、本发明通过对结直肠癌个体和健康个体的粪便样本中微生物的丰度进行差异分析和比较,标志物在结直肠癌患者的粪便样本中的含量显著高于在健康个体的粪便样本中的含量,且具有统计学意义,能够较为准确地确定个体是否患有结直肠癌,具有非侵入性辅助诊断结直肠癌的作用,仅需粪便即可检测,患者耐受性强。
28.第二、本发明在crc早期即可检测,一方面,可以早发现早诊断crc病例,可以显著提高crc患者的生存期;另一方面,可以有效节约医疗资源,从而更好地配置稀缺医疗资源。
29.第三、本发明所设计的单物种预测模型包括16种古菌、26种细菌、24种真菌和102种病毒,纳入物种广泛且量多,准确性高。
附图说明
30.图1为本发明的四界中crc的总体微生物丰度示意图。
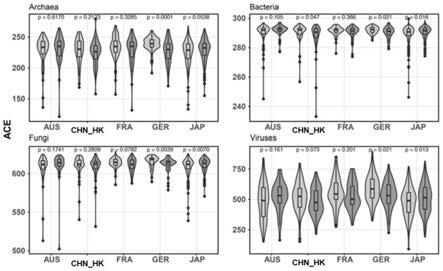
31.图2为本发明的四界中古菌、细菌、真菌和病毒丰度示意图。
32.图3为本发明的四界中crc与对照样本的肠道微生物组组成示意图。
33.图4为本发明的四界中微生物种类变化示意图。
34.图5为本发明的四界中古菌、细菌和病毒种类变化示意图。
35.图6为本发明的古菌、细菌、真菌和病毒的auroc值示意图。
36.图7为本发明每一个单一界别物种构建的分类模型在另一组独立人群队列中预测能力的表现图。
37.图8为本发明的两界中细菌和真菌的组合示意图。
38.图9为本发明的三界中细菌、真菌、古菌的预测示意图。
39.图10为本发明的四界中细菌、真菌、古菌和病毒的预测示意图。
40.图11为本发明的细菌-真菌模型中预测示意图。
41.图12为本发明的细菌和真菌组合特征构建的模型预测能力表现图。
42.图13为本发明运用内部随机森林基尼重要性方法通过预测能力表现交互验证评估细菌
‑ꢀ
真菌组合特征的重要性图。
43.图14为本发明的验证队列中非crc病例样本和相应对照样本auc比较示意图。
具体实施方式
44.本发明提供了一种结直肠癌的微生物标志物及其应用。
45.本发明首次发现,在不同地域人群中,crc患者和健康对照组之间除了细菌,在真菌、古菌和病毒也存在差异。目前尚未见有报道用于临床或关于结直肠癌筛查的四大微生物类群菌群谱,本发明首次进行了大规模的人群分析,并得到了囊括四大菌群的综合性、系统性、整合性的微生物组图谱,而且能更准确地辅助诊断结直肠癌。
46.因此,本发明主要涉及crc的综合致癌微生物群,包括细菌、真菌、古菌和病毒。一方面,本发明在9个跨地域不同队列人群中测试了单物种诊断crc的可预测性,并确定了一组微生物特征,这些特征可作为crc特异性诊断的生物标志物,具有广泛的推广价值;因此本发明首次纳入来自9个队列的1368个大样本量人群,并且数据经过验证队列的验证。另一方面,本发明还发现,四个物种之间的组合诊断模型,其受试者工作特征曲线下面积(auroc)要显著优于单个模型,尤其是细菌和真菌的组合诊断模型达到了最优的诊断效果。
47.所以,本发明的结直肠癌微生物标志物能够较为准确地确定个体是否患有结直肠癌,具有非侵入性辅助诊断结直肠癌的作用。
48.具体地,本发明中涉及9个队列共计1368个样本,包括5个发现队列和4个验证队列。
49.1.发现队列的建立:
50.5个发现队列来自5个国家(奥地利、法国、德国、中国(香港)和日本)的491名crc 受试者和494名无瘤对照者,将这5个队列分别记为aus、fra、ger、chn_hk和jap,数据均来自公开数据库。
51.其中,aus:https://www.ebi.ac.uk/ena/browser/view/prjeb7774
52.fra:https://www.ebi.ac.uk/ena/browser/view/prjeb6070
53.ger:https://www.ebi.ac.uk/ena/browser/view/prjeb27928
54.chn_hk:https://www.ebi.ac.uk/ena/browser/view/prjeb10878
55.jap:https://www.ncbi.nlm.nih.gov/sra/?term=dra006684
56.1.1四界中与crc相关微生物的单变量分析:
57.a.crc的总体微生物丰度呈下降趋势(如图1所示)。
58.b.同样的下降趋势在古生菌、细菌、真菌和病毒微生物组也有观察到(如图2所示)。
59.c.在门水平上,本发明发现crc与对照样本在的四界中的肠道微生物组组成不同(如图3 所示)。
60.d.在种水平上确定crc患者的特定微生物种类变化,不同的微生物种类在组间差异很大,只有少数种类在组间表现出一致的趋势,其中96种细菌、56种真菌、25种古细菌和158 种病毒在crc组和对照组的丰度存在差异,42种细菌、37种真菌、56种病毒在crc患者中丰度升高(如图4和图5所示)。
61.1.2基于单一界物种的crc微生物诊断模型的构建:诊断模型的能力由受试者工作特征曲线下面积(auroc)判断,用来自各界的物种构建分类模型,并根据它们对总体微生物丰度的贡献,依次选择信息最丰富的物种。最终,我们确定了13种古生菌、26种细菌、24种真菌和102种病毒。
62.a.除了法国(fra)和德国(ger)队列,细菌模型在所有队列中显示出最强的诊断crc的能力(auroc:0.73-0.88)。本发明的真菌生物标志物模型显示出卓越的区分能力(最大auroc 为0.88,最小auroc为0.65),特别是在fra(0.87vs 0.85auroc)和ger(0.85vs 0.80 auroc)数据集中,它表现得比细菌模型更好。同时,带有古菌平均auroc(avg auroc) 为0.72和病毒(avg auroc)为0.75生物标记的模型的auroc值略低,均如图6所示。
63.b.为了评估上述任何一单界生物标志物是否在crc中具有普遍性并克服地理异质性,我们进行了队列间转移分析和loco分析。在队列到队列的迁移分析中,该模型使用一个单队列数据集作为训练数据,随后在不同队列中验证作为测试数据。总体而言,与上述队列内模型相比,队列间转移验证的auroc评分降低,并显示出队列间的高可变性(如图6所示)。
64.c.为了进一步减少在单个数据集上训练的限制,我们对模型进行了locl分析,分别在合并的、四个队列的数据集上训练模型,并在分别被排除的队列上验证模型。与队列间迁移分析相比,获得的auroc值有所增加,这可能是因为训练数据集的规模更大。此外,我们在数据集内模型中观察到类似的趋势,细菌模型获得最高的中位auroc(0.77),其次是真菌模型(中位auroc=0.75)、病毒模型(中位auroc=0.74)和古菌模型(中位auroc=0.71)。总的来说,这些多界生物标志物在具有不同地理背景的队列中均显示了对crc的无偏预测能力(如图6所示)。
65.2.验证队列的建立:4个验证队列来自3个国家(中国(上海)、意大利和美国)的193例 crc患者和190例对照组,将这4个队列分别记为chn_sh、itaa、itab和usa。usa、 itaa、itab来自公开数据库。
66.其中,usa:https://www.ebi.ac.uk/ena/browser/view/prjeb12449
67.itaa:https://www.ncbi.nlm.nih.gov/sra/?term=srp136711
68.itab:https://www.ncbi.nlm.nih.gov/sra/?term=srp136711
69.chn_sh为本发明新建立的队列。
70.2.1chn_sh队列的建立包括以下三步:
71.a.粪便样本的收集;
72.b.样本粪便dna的提取;
73.c.测序和序列分析。
74.2.2使用独立的数据集进行额外的外部验证,以评估预筛选模型,避免过于乐观的模型准确性报告。所有的模型准确地检测到crc患者,每个独立单一界物种模型显示更高的检测精度(细菌最大(auroc为0.92)和古菌最低(auroc为0.86)),如图7所示。
75.3.基于组合多界物种特征的crc诊断模型改进:
76.通过对四大类群微生物组之间的互作分析获得了预测结直肠癌的更准确的标记物-微生物互作图谱(包括两两互作、三者互作、四者互作),其中细菌-真菌互作对的预测能力更强、准确性最高,例如fusobacterium nucleatum和aspergillus ochraceoroseus组合后可以显著刺激结直肠癌细胞增殖、促进细胞黏附并上调癌症相关基因表达等,此外还包括 clostridiumbolteae-candida pseudohaemulonis,clostridiumbolteae-nadsonia fulvescens互作,可以作为结直肠癌筛查以及结直肠癌发病机制的重要指标,极其有潜力成为结直肠癌筛查和早期诊断的非侵入性生物标志物。
77.3.1探索多界生物标志物组合的可预测性。
78.a.用两界生物标志物构建诊断模型。与单界诊断模型相比,双界生物标志物的auroc 值显著提高,均在0.81以上,高于大多数单界生物标志物的auroc值。其中,结合细菌和古生菌生物标志物的队列内模型的平均auroc为0.84,高于任何单界模型(细菌的auroc 为0.83,古生菌的auroc为0.72)。在其他两界生物标志物上也观察到类似的改善。值得注意的是,结合细菌和真菌生物标志物的模型的预测价值优于其他组合,达到平均 auroc=0.86。不同队列的auroc得分分别为0.93(aus)、0.86(chn_hk)、0.89(fra)、0.88(ger)和0.75(jap),如图8所示。
79.b.用三界生物标志物构建诊断模型。基于三界特征组合的模型预测值略高。细菌-真菌
‑ꢀ
古菌模型的表现优于其他模型,平均auroc(队列内)为0.85,高于任何单一界模型,但低于最优的两界模型。(a=古生菌;b=细菌;f=真菌;v=病毒),如图9所示。
80.c.用四界生物标志物构建诊断模型。基于四界生物标志物组合的模型的auroc也没有进一步改善(avg auroc=0.84),如图10所示。
81.3.2确定细菌-真菌模型预测价值的基本特征。
82.a.在细菌-真菌模型中,a.niger(黑曲霉)丰度是一个平均排名最高的重要物种。其次,两种细菌,f.nucleatum和p.micra,是这些模型预测价值的第二和第三重要贡献者。同时, talaromycesislandicus,a.rambellii,sistotremastrumsuecicum,trichophytonmentagrophytes等真菌种类以及gemellamorbillorum、p.ascharolytica、ruminococcusbicirculans等是最重要的前十贡献值者。此外,产丁酸细菌roseburiacoccinalis和butyricimonasfaecalis,致病菌 bacteroidescaccae和dialisterpneummosintes,真菌种lipomycesstarkeyi,saccharomycescerevisiae和a.ochraceoroseus也在细菌-真菌模型中具有较高的平均等级。因此,这些特征排名分析强调了结合来自多个界的特征的必要性,特别是来自细菌和真菌界的特征,以最大化预测价值,如图11所示。
83.b.根据细菌和真菌丰度的排名依次添加物种来建立最小物种数的生物标记物panel。添加排名前13个物种后(如下),平均auroc曲线开始最大化,auroc为0.84(如图13所示)。构建的模型在单个队列中也取得了良好的表现。此外,除jap(auroc=0.71)外,所有队列模型识别crc样本的准确率均高于0.84,aus队列的预测力最高,auroc为0.92,模型还显示出了跨队列的强可转移性(如图12中a所示),并且对独立验证数据集具有更高的预测值(如图12中b所示)。因此,本发明证明了13个来自细菌和真菌界的生物标志物作为基于粪便的无创crc预筛查工具的适用性。
84.13个物种具体名称:
85.1.a.niger(fungi)
86.2.f.nucleatum(bacteria)
87.3.p.micra(bacteria)
88.4.t.islandicus(fungi)
89.5.a.rambellii(fungi)
90.6.g.morbillorum(bacteria)
91.7.p.asaccharolytica(bacteria)
92.8.s.suecicum(fungi)
93.9.r.bicirculans(bacteria)
94.10.t.mentagrophytes(fungi)
95.11.r.intestinalis(bacteria)
96.12.b.caccae(bacteria)
97.13.d.pneumosintes(bacteria)
98.3.3细菌-真菌生物标志物在crc预测模型中的特异性:
99.由于不同疾病的共同微生物群改变,有必要验证所识别的微生物标志物的疾病特异性,并确保crc诊断模型的低假阳性率。因此,我们评估鉴别出的细菌和真菌特征组合对crc 预测是否具有特异性。一些非crc疾病数据集被评估,包括胃肠道疾病ibd(uc和cd)和非胃肠道疾病如2型糖尿病(t2d)、肝硬化(lc)和帕金森病(pd)。我们从每个外部对照组和病例 (疾病状态)组随机抽取样本,并将它们添加到独立验证队列的对照类中,以消除不同数据集之间的批处理效应。通过加入外部非crc病例样本和相应对照样本后的auc比较,如图14 所示,我们发现chn-sh、itaa和usa队列中cd和uc的auc均略有下降,而itab队列的准确率有所提高。这一数据表明,来自这两种胃肠道疾病的样本对我们的crc预测模型的影响有限。对于t2d、肝硬化和帕金森病,在相同的四个验证队列中,仅观察到预测准确性的微小变化,表明区分病例和外部对照的能力相似。总的来说,模型保持了很高的预测精度(平均auc》0.80),表明我们的细菌-真菌模型的综合特征与其他微生物群系相关疾病无关,因此对crc具有特异性。
100.其中,在单界微生物标志物的预测模型中,26种细菌、24种真菌、16种古菌以及102 种病毒在结直肠癌组织中显著富集,可以作为潜在的预测结直肠癌的微生物标志物。
101.实际上,细菌单物种预测模型包括26种细菌,具体如下:
102.1.fusobacteriumnucleatum
103.2.parvimonasmicra
104.3.gemellamorbillorum
105.4.porphyromonasasaccharolytica
106.5.ruminococcusbicirculans
107.6.roseburiaintestinalis
108.7.bacteroidescaccae
109.8.dialisterpneumosintes
110.9.lachnospiraceae bacterium km106-2
111.10.pseudobutyrivibrioxylanivorans
112.11.streptococcus thermophiles
113.12.lactobacillus ruminis
114.13.butyricimonasfaecalis
115.14.odoribactersplanchnicus
116.15.ruthenibacteriumlactatiformans
117.16.streptococcus anginosus
118.17.faecalibacteriumprausnitzii
119.18.fusobacteriumnecrophorum
120.19.flavonifractorplautii
121.20.bifidobacteriumpseudocatenulatum
122.21.streptococcus oralis
123.22.alistipesfinegoldii
124.23.bacteroidescellulosilyticus
125.24.clostridiales bacterium ccna10
126.25.mogibacteriumdiversum
127.26.prevotellascopos
128.真菌单物种预测模型包括24种真菌,具体如下:
129.1.aspergillus niger
130.2.talaromycesislandicus
131.3.aspergillus rambellii
132.4.sistotremastrumsuecicum
133.5.trichophytonmentagrophytes
134.6.lipomycesstarkeyi
135.7.saccharomyces cerevisiae
136.8.penicilliumbrasilianum
137.9.rhizophagusirregularis
138.10.lachanceadasiensis
139.11.aspergillus ochraceoroseus
140.12.nosemabombycis
141.13.blastomycesparvus
142.14.mucorambiguus
143.15.malasseziavespertilionis
144.16.hypsizygusmarmoreus
145.17.sclerotiniasclerotiorum
146.18.nadsoniafulvescens
147.19.absidiaglauca
148.20.erysiphepulchra
149.21.tilletia caries
150.22.aspergillus violaceofuscus
151.23.aspergillus nomiae
152.24.rhizopusmicrospores
153.古菌单物种预测模型包括16种古菌,具体如下
154.1.thermosphaera aggregans
155.2.sulfodiicoccus acidiphilus
156.3.sulfolobus acidocaldarius
157.4.sulfuracidifex tepidarius
158.5.sulfurisphaera tokodaii
159.6.thermofilum uzonense
160.7.pyrobaculum arsenaticum
161.8.pyrobaculum neutrophilum
162.9.halorubrum lacusprofundi
163.10.methanobrevibacter smithii
164.11.methanosphaera sp.bms
165.12.methanothermobacter sp.kepco-1
166.13.methanococcoides methylutens
167.14.pyrococcus horikoshii
168.15.thermococcus gorgonarius
169.16.thermococcus kodakarensis
170.病毒单物种预测模型包括102种病毒,具体如下:
171.1.human mastadenovirus b
172.2.simian mastadenovirus e
173.3.torque teno canis virus
174.4.spodoptera frugiperda ascovirus 1a
175.5.perigonia lusca nucleopolyhedrovirus
176.6.spodoptera littoralis nucleopolyhedrovirus
177.7.adoxophyes orana granulovirus
178.8.acidianus tailed spindle virus
179.9.tea plant line pattern virus
180.10.providence virus
181.11.tea plant necrotic ring blotch virus
182.12.rosellinia necatrix megabirnavirus 2-w8
183.13.moumouvirus
184.14.rhizoctonia solani dsrna virus 2
185.15.paramecium bursaria chlorella virus a1
186.16.aureococcus anophagefferens virus
187.17.african eggplant mosaic virus
188.18.pepper severe mosaic virus
189.19.seal parapoxvirus
190.20.bean 58058 virus
191.21.solenopsis invicta virus 3
192.22.thermus virus in93
193.23.pandoravirus macleodensis
194.24.pandoravirus salinus
195.25.tadarida brasiliensis polyomavirus 1
196.26.xipapillomavirus 2
197.27.parus major densovirus
198.28.red-crowned crane parvovirus
199.29.varroa mite associated genomovirus 1
200.30.dickeya virus limestone
201.31.dickeya virus rc201
202.32.pantoea virus limelight
203.33.citrobacter phage cr44b
204.34.enterobacteria phage 285p
205.35.synechococcus phage s-rip1
206.36.enterobacter virus ecl1
207.37.shigella virus psf1
208.38.klebsiella virus pkp126
209.39.bacillus virus bc431
210.40.staphylococcus virus mce2014
211.41.listeria virus lmta148
212.42.bacillus virus mater
213.43.mycobacterium virus lukilu
214.44.pandoravirus macleodensis
215.45.pandoravirus salinus
216.46.tadarida brasiliensis polyomavirus 2
217.47.xipapillomavirus 3
218.48.parus major densovirus
219.49.red-crowned crane parvovirus
220.50.varroa mite associated genomovirus 2
221.51.dickeya virus limestone
222.52.dickeya virus rc2015
223.53.pantoea virus limelight
224.54.citrobacter phage cr45b
225.55.enterobacteria phage 286p
226.56.synechococcus phage s-rip2
227.57.enterobacter virus ecl2
228.58.shigella virus psf2
229.59.pandoravirus macleodensis
230.60.pandoravirus salinus
231.61.tadarida brasiliensis polyomavirus 2
232.62.xipapillomavirus 3
233.63.parus major densovirus
234.64.red-crowned crane parvovirus
235.65.varroa mite associated genomovirus 2
236.66.dickeya virus limestone
237.67.dickeya virus rc2015
238.68.pantoea virus limelight
239.69.citrobacter phage cr45b
240.70.enterobacteria phage 286p
241.71.synechococcus phage s-rip2
242.72.enterobacter virus ecl2
243.73.shigella virus psf2
244.74.klebsiella virus pkp127
245.75.bacillus virus bc432
246.76.staphylococcus virus mce2015
247.77.listeria virus lmta149
248.78.bacillus virus mater
249.79.mycobacterium virus lukilu
250.80.pandoravirus macleodensis
251.81.pandoravirus salinus
252.82.tadarida brasiliensis polyomavirus 2
253.83.xipapillomavirus 3
254.84.parus major densovirus
255.85.red-crowned crane parvovirus
256.86.varroa mite associated genomovirus 2
257.87.dickeya virus limestone
258.88.dickeya virus rc2015
259.89.pantoea virus limelight
260.90.citrobacter phage cr45b
261.91.enterobacteria phage 286p
262.92.synechococcus phage s-rip2
263.93.enterobacter virus ecl2
264.94.shigella virus psf2
265.95.klebsiella virus pkp127
266.96.bacillus virus bc432
267.97.staphylococcus virus mce2015
268.98.listeria virus lmta149
269.99.bacillus virus mater
270.100.mycobacterium virus lukilu
271.101.gordonia virus oneup
272.102.arthrobacter virus tank
273.以下结合实施例对本发明作进一步的说明。
274.实施例:
275.本实施例的验证队列的构建过程包括:
678.doi:10.1038/s41591-019-0405-7.epub2019apr1.erratumin:natmed.2019dec;25(12):1948.pmid:30936548.
294.wirbelj,pylpt,kartale,zychk,kashania,milanesea,fleckjs,voigtay,pallejaa,ponnudurair,sunagawas,coelholp,schrotz-kingp,vogtmanne,habermannn,nim
é
use,thomasam,manghip,gandinis,serranod,mizutanis,shiromah,shibas,shibatat,yachidas,yamadat,waldronl,naccaratia,segatan,sinhar,ulrichcm,brennerh,arumugamm,borkp,zellerg.meta-analysisoffecalmetagenomesrevealsglobalmicrobialsignaturesthatarespecificforcolorectalcancer.natmed.2019apr;25(4):679-689.doi:10.1038/s41591-019-0406-6.epub2019apr1.pmid:30936547;pmcid:pmc7984229.
295.日本队列网址:https://www.ncbi.nlm.nih.gov/sra/?term=dra006684
296.上述对实施例的描述是为了便于该技术领域的普通技术人员能理解和使用本发明。熟悉本领域技术人员显然可以容易的对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中,而不必经过创造性的劳动。因此,本发明不限于上述实施例。本领域技术人员根据本发明的原理,不脱离本发明的范畴所做出的改进和修改都应该在本发明的保护范围之内。