1.本发明属于化学技术领域,具体涉及并四苯与蒽苯基桥连聚合体及其制备方法。
背景技术:
2.并四苯和蒽作为一种常见的有机半导体材料,具有可调的光电性质、较高的电荷迁移率,在有机场效应晶体管、有机光伏等领域具有潜在的应用。研究其合成物质对于拓宽潜在领域和应用有着重要的意义。
技术实现要素:
3.本发明的目的是针对现有的问题,提供了并四苯与蒽苯基桥连聚合体及其制备方法,通过苯基桥连合成并四苯与蒽苯基桥连聚合体,拓宽了光谱,在其中出现了新的光电性质,并在其中一个化合物上引入羧基提高了其稳定性,并使其具有了两亲性。
4.本发明是通过以下技术方案实现的:
5.并四苯与蒽苯基桥连聚合体,该物质为下述式(i)或式(ii)的化合物:
[0006][0007]
[0008]
2.并四苯与蒽苯基桥连聚合体的制备方法,其特征在于,包括以下合成步骤:
[0009][0010]
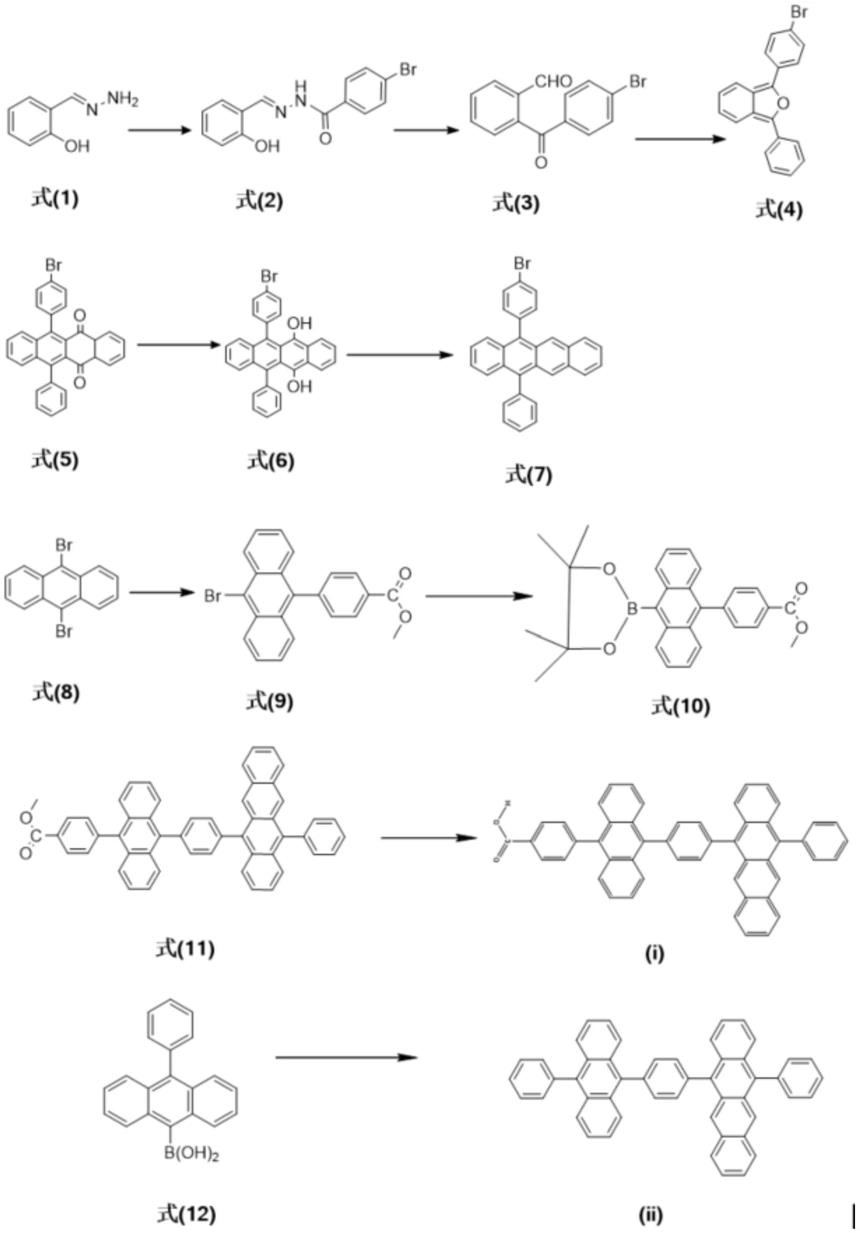
a.将式(1)化合物加入到盛有无水四氢呋喃的三口瓶中,移取二异丙基乙胺加入上述三口瓶中,室温条件下搅拌,称取对溴苯甲酰氯加入盛有四氢呋喃的恒压低液漏斗中,然后缓慢滴入到三口瓶中,反应3h,停止反应,将反应液倒入水中,抽滤,水洗3次,取滤饼,真空干燥,得式(2)化合物;
[0011]
b.称取式(2)化合物加入到盛有四氢呋喃的三口瓶中,搅拌,降温至0℃,然后称取四乙酸铅溶于盛有四氢呋喃的恒压低液漏斗中,缓慢滴入上述三口瓶,反应5h,停止反应,减压蒸干四氢呋喃,加入乙酸乙酯,抽滤,取有机相,减压蒸干乙酸乙酯,得粗产物,柱层析色谱提纯得到式(3)化合物;
[0012]
c.称取式(3)化合物,加入盛有四氢呋喃的三口瓶中,搅拌,降温至0℃,移取苯基溴化镁加入到恒压低液漏斗中,然后缓慢滴入到三口瓶,继续反应8h,然后加入稀盐酸,反应升至室温,继续反应1h,停止反应,萃取,收集有机相,减压蒸干有机溶剂,柱层析色谱提纯得式(4)化合物;
[0013]
d.称取萘醌溶于盛有二氯甲烷的三口瓶中,室温条件下,氮气保护,将式(4)化合物分三次加入上述三口瓶,搅拌反应16h,然后冷却至零下78℃,移取三溴化硼加入到盛有二氯甲烷的恒压低液漏斗中,然后缓慢滴入上述反应,继续反应2h,然后升温至室温,反应5h,加热回流6h,停止反应,反应液倒入大量的水中,萃取,收集有机相,减压蒸干有机溶剂,柱层析色谱提纯得到式(5)化合物;
[0014]
e.称取式(5)化合物,将硼氢化钠溶于乙醇,升温至75℃,反应4h,停止反应,萃取,收集有机相,减压蒸干有机溶剂,得式(6)化合物;
[0015]
f.称取式(6)化合物、氯化亚锡加入盛有四氢呋喃的三口瓶中,氮气保护,室温反应6h,停止反应,萃取,收集有机相,减压蒸干溶剂,粗产品柱层析色谱提纯得式(7)化合物;
[0016]
g.将9,10-二溴蒽,4-甲氧羰基苯硼酸,四(三苯基)磷钯,dpehos和k2co3的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30min的甲苯和乙醇,加热至95℃并在黑暗中反应7.5h,反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷和水的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物,用硅胶柱色谱纯化得到化合物(9);
[0017]
h.将化合物(9)、联硼酸频那醇酯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯和醋酸钾的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30min的dmf,加热至95℃并在黑暗中反应18h,反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷和水的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物,用硅胶柱色谱纯化得到化合物(10);
[0018]
i.将化合物(10)、化合物(7)、四(三苯基)磷钯、dpehos和k2co3的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30min的甲苯和乙醇,加热至95℃并在黑暗中反应8h,反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷和水的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物,用硅胶柱色谱纯化得到化合物(11)橙色固体;
[0019]
j.将化合物(11)、koh的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30min的thf、甲醇和h2o加热至60℃并在黑暗中反应8h,反应结束后,等混合物冷却至室温,然后加入hcl溶液酸化5h,加入碳酸氢钠中和,再将溶液转移到含有二氯甲烷和水的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物,用硅胶柱色谱纯化得到化合物(
ⅰ
);
[0020]
k.将10-苯基-9-蒽硼酸、化合物(7)、四(三苯基)磷钯、dpehos和k2co3的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡
30min的甲苯和乙醇,加热至95℃并在黑暗中反应8h,反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷和水的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物,用硅胶柱色谱纯化得到化合物(
ⅱ
)橙色固体。
[0021]
进一步的,步骤b中所述的洗脱剂为二氯甲烷。
[0022]
进一步的,步骤c中所述的洗脱剂是正己烷。
[0023]
进一步的,步骤d中所述的洗脱剂是二氯甲烷。
[0024]
进一步的,步骤e中所述的洗脱剂是石油醚和三氯甲烷对应体积比为7:1的混合溶剂。
[0025]
进一步的,步骤g中所述的洗脱剂是二氯甲烷和正己烷对应体积比为5:3的混合溶剂。
[0026]
进一步的,步骤h中所述的洗脱剂是二氯甲烷和正己烷对应体积比为3:2的混合溶剂。
[0027]
进一步的,步骤i中所述的洗脱剂是二氯甲烷和正己烷对应体积比为1:1的混合溶剂。
[0028]
进一步的,步骤j中所述的洗脱剂是二氯甲烷和甲醇对应体积比为10:1的混合溶剂;
[0029]
步骤k中所述的洗脱剂是二氯甲烷和正己烷对应体积比为1:1的混合溶剂。
[0030]
本发明相比现有技术具有以下优点:
[0031]
本发明通过苯基桥连合成并四苯与蒽苯基桥连聚合体,可以使蒽与并四苯发生能量传递,使其在一些光电器件中表现出一些新颖的光电特性;另外,合成了带有羧基四苯与蒽苯基桥连聚合体使分子具有两亲性,对于其构成自组装纳米颗粒时分子具有取向性,更有序的排列,此外羧基也为其与量子点等无机材料的复合提供基础。
具体实施方式
[0032]
实施例1
[0033]
并四苯与蒽苯基桥连聚合体,其化学结构式如式(
ⅰ
)或(
ⅱ
)所示:
[0034][0035]
并四苯与蒽苯基桥连聚合体的制备方法,包括以下合成步骤:
[0036][0037]
a.称取1.36g式(1)化合物加入到盛有100ml无水四氢呋喃的三口瓶中,移取1.64ml二异丙基乙胺加入上述三口瓶中,室温条件下搅拌,称取2.2g的对溴苯甲酰氯加入盛有50ml四氢呋喃的恒压低液漏斗中,然后缓慢滴入到三口瓶中,反应3小时,停止反应,将反应液倒入大量的水中,抽滤,水洗3次,取滤饼,真空干燥,得式(2)化合物(2.83g),产率89%;
[0038]
b.称取式(2)化合物3.18g加入到盛有100ml四氢呋喃的三口瓶中,搅拌,降温至0℃,然后称取4.43g四乙酸铅溶于盛有100ml四氢呋喃的恒压低液漏斗中,缓慢滴入上述三口瓶,反应5小时,停止反应,减压蒸干四氢呋喃,加入乙酸乙酯,抽滤,取有机相,减压蒸干乙酸乙酯,得粗产物,柱层析色谱提纯得到式(3)化合物(2.35g,产率:81%),洗脱剂为二氯甲烷;
[0039]
c.称取1g式(3)化合物,加入盛有100ml四氢呋喃的三口瓶中,搅拌,降温至0℃,移取7ml苯基溴化镁(1mol/l)加入到恒压低液漏斗中,然后缓慢滴入到三口瓶,继续反应8小时,然后加入17.5ml的稀盐酸(4mol/l),反应升至室温,继续反应1小时,停止反应,萃取,收集有机相,减压蒸干有机溶剂,柱层析色谱提纯得式(4)化合物纯品,洗脱剂是正己烷;
[0040]
d.称取0.45g萘醌溶于盛有100ml二氯甲烷的三口瓶中,室温条件下,氮气保护,将1g式(4)化合物分三次加入上述三口瓶,搅拌反应16小时,然后冷却至零下78℃,移取3ml三溴化硼(1mol/l)加入到盛有10ml二氯甲烷的恒压低液漏斗中,然后缓慢滴入上述反应,继续反应2小时,然后升温至室温,反应5小时,加热回流6小时,停止反应,反应液倒入大量的水中,萃取,收集有机相,减压蒸干有机溶剂,柱层析色谱提纯得到式(5)化合物,洗脱剂是二氯甲烷;
[0041]
e.称取122mg式(5)化合物,19mg硼氢化钠溶于25ml乙醇,升温至75℃,反应4小时,停止反应,萃取,收集有机相,减压蒸干有机溶剂,得式(6)化合物;
[0042]
f.称取98mg式(6)化合物、76mg氯化亚锡加入盛有20ml四氢呋喃的三口瓶中,氮气保护,室温反应6小时,停止反应,萃取,收集有机相,减压蒸干溶剂,粗产品柱层析色谱提纯得式(7)化合物,洗脱剂是石油醚和三氯甲烷的混合溶剂(体积比为7:1);
[0043]
g.9,10-二溴蒽(336mg,1mmol),4-甲氧羰基苯硼酸(179.9mg,1mmol),四(三苯基)磷钯(57.8mg,0.05mmol),dpehos(53mg,0.1mmol)和k2co3(691mg,5mmol)的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30分钟的甲苯(24ml)和乙醇(2ml)。加热至95℃并在黑暗中反应7.5小时。反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷(50ml)和水(100ml)的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物用硅胶柱色谱纯化(二氯甲烷/正己烷=5/3),得到化合物(9),235.6mg,产率60%;
[0044]
h.化合物(9)(390mg,1mmol),联硼酸频那醇酯(507.5mg,2mmol),[1,1'-双(二苯基膦基)二茂铁]二氯化钯(75mg,0.1mmol)和醋酸钾(2g,20mmol)的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30分钟的dmf(50ml)。加热至95℃并在黑暗中反应18小时。反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷(50ml)和水(100ml)的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物用硅胶柱色谱纯化(二氯甲烷/正己烷=3/2),得到化合物(10),284.7mg,产率65%;
[0045]
i.将化合物(10)(87.6mg,0.2mmol),化合物(7)(91.8mg,0.2mmol),四(三苯基)磷钯(11.56mg,0.01mmol),dpehos(53mg,0.1mmol)和k2co3(140mg,1mmol)的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30分钟的甲苯(24ml)和乙醇(2ml)。加热至95℃并在黑暗中反应8小时。反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷(50ml)和水(100ml)的分液漏斗中,分离有机层,
用无水mgso4干燥所得粗产物用硅胶柱色谱纯化(二氯甲烷/正己烷=1/1),得到化合物(11)橙色固体,110.4mg,产率80%;
[0046]
j.将化合物(11)(690mg,1mmol),koh(8g,20mmol)的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30分钟的thf(80ml),甲醇(80ml)和h2o(30ml)。加热至60℃并在黑暗中反应8小时。反应结束后,等混合物冷却至室温,然后加入40ml 1mol/l hcl溶液酸化5h,加入碳酸氢钠中和,再将溶液转移到含有二氯甲烷(50ml)和水(100ml)的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物用硅胶柱色谱纯化(二氯甲烷/甲醇=10/1),得到化合物(
ⅰ
),608.4mg,产率90%;
[0047]
k.10-苯基-9-蒽硼酸(59.6mg,0.2mmol),化合物(7)(91.8mg,0.2mmol),四(三苯基)磷钯(11.56mg,0.01mmol),dpehos(53mg,0.1mmol)和k2co3(140mg,1mmol)的混合物加入至干燥的圆底烧瓶,抽真空,抽换氩气三次以排除反应装置中的氧气,然后加入氮气鼓泡30分钟的甲苯(24ml)和乙醇(2ml)。加热至95℃并在黑暗中反应8小时。反应结束后,等混合物冷却至室温,然后将溶液转移到含有二氯甲烷(50ml)和水(100ml)的分液漏斗中,分离有机层,用无水mgso4干燥所得粗产物用硅胶柱色谱纯化(二氯甲烷/正己烷=1/1),得到化合物(
ⅱ
)橙色固体,110.4mg,产率80%。
[0048]
本发明公开了一类并四苯与蒽聚集体及其制备方法,首先制得卤代并四苯;和修饰有硼酸酯基的蒽其;最后经过suzuki偶合反应制得并四苯与蒽聚集体。本发明的有益效果是可以拓宽并四苯的吸收光谱,使其在一些光电器件中表现出一些新颖的光电特性;构建宽光谱吸收的单线态裂分体系。
[0049]
最后应说明的是:以上所述仅为本发明的优选内容而已,并不用于限制本发明,尽管参照前述内容对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各内容所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。