1.本发明涉及人类遗传学和内科心血管技术领域,尤其涉及先天性心脏病突变基因及其应用。
背景技术:
2.先天性心脏病(congenital heart disease,chd)是儿科常见的疾病,是胎儿时期心脏及大血管发育异常而致的先天畸形,是小儿最常见的心脏病,发病率在活产婴儿中为4-8
‰
,若包括出生前即死亡的胎儿。发生率更高。
3.现已发现约有899多种临床综合征伴有chd,临床较为常见的有alagille综合征、charge联合征、holt-oram综合征(hos)、noonan综合征、turner综合征、vacterl联合征、vacterl综合征等,其中,holt-oram综合征的女性比男性严重,同一家族表型严重度不同,上肢异常很轻微的患者需x光片才能辨认,发病率1/100 000。85-95%的患者。临床主要表现为:房间隔缺损(第二孔型),室间隔缺损,左心发育不全综合症,动脉导管未闭,胸大肌缺如,漏斗胸或鸡胸,脊椎畸形,胸椎侧凸,拇指缺如,拇指裂,拇指三指节畸形,腕骨异常,上肢短肢畸形,桡尺异常,牵连不对称。
4.holt-oram综合征为常染色体显性遗传,外显率100%,表型多变,致病基因tbx5的无义突变导致上肢和心脏同时畸形,错义突变导致其它表型,30-40%的患者存在tbx5突变。tbx5基因是一个系统发育保守的基因家族成员,该家族共享一个dna结合结构域,即t-box。t-box基因编码调控发育过程的转录因子。编码蛋白可能在心脏发育和肢体识别中发挥作用。
5.目前仍存在大量未知的tbx5基因突变位点,进一步发现新的tbx5基因的突变位点对于研究holt-oram综合征的发病机制,对holt-oram综合征的早期诊断,或者辅助临床判断具有重要意义。
技术实现要素:
6.本发明的目的是针对holt-oram综合征,提供一种先天性心脏病突变基因及其应用。
7.本发明提供的技术方案如下:
8.一、本发明提供先天性心脏病突变基因,与野生型tbx5基因编码dna的参考序列相比,先天性心脏病突变基因的核苷酸序列为seq id no:3;在基因组位置chr12:114837380处,碱基c突变为碱基g;参考基因组版本是grch37。
9.二、本发明还提供了上述先天性心脏病突变基因在制备检测试剂盒中的应用。
10.优选地,所述检测试剂盒包括引物seq id no:1和seq id no:2。
11.优选地,所述检测试剂盒还包括taq dna聚合酶和pcr缓冲液。
12.三、本发明的原理和有益效果在于:
13.本发明公开的突变基因可以作为临床辅助诊断的生物标志物,对holt-oram综合
征的早期诊断,或者辅助临床判断具有重要意义;基于突变基因开发的检测试剂盒,可以检测出具有先天性心脏病突变基因的患者,为受试者提供优生优育指导和遗传咨询,减少患儿出生。
附图说明
14.图1为实施例3的家系图;
15.图2为实施例3中先证者、先证者母亲等人的sanger测序图;
16.图3为实施例3家系中先证者父亲、先证者外婆等人的sanger测序图。
具体实施方式
17.下面通过具体实施方式进一步详细说明:
18.实施例1-先天性心脏病突变基因
19.先天性心脏病突变基因,具体突变如下表1所示:
20.表1先天性心脏病突变基因的具体检测结果
21.基因基因组位置转录本号碱基改变氨基酸改变参考基因组版本外显子号tbx5chr12:114837380nm_080717c.150c》gp.tyr50tergrch37/hg19exon3
22.(1)在基因组位置chr12:114837373-chr12:114837421处,野生型tbx5基因的序列为:
23.ttacaaagtgaaggtgacgggccttaatcccaaaacgaagtaattcttc,为野生型tbx5基因在基因组chr12:114837380处的碱基。
24.在对应基因组位置处,先天性心脏病突变基因的序列为:
25.ttacaaagtgaaggtgacgggccttaatcccaaaacgaagtaattcttc,为突变基因在基因组chr12:114837380处的碱基。
26.(2)野生型tbx5基因编码dna的参考序列为:
27.atggagggaatcaaagtgtttctccatgaaagagaactgtggctaaaattccacgaagtgggcacggaaatgatcataaccaaggctggaaggcggatgtttcccagttacaaagtgaaggtgacgggccttaatcccaaaacgaagtaattcttctcatggacattgtacctgccgacgatcacagatacaaattcgcagataataaatggtctgtgacgggcaaagctgagcccgccatgcctggccgcctgtacgtgcacccagactcccccgccaccggggcgcattggatgaggcagctcgtctccttccagaaactcaagctcaccaacaaccacctggacccatttgggcatattattctaaattccatgcacaaataccagcctagattacacatcgtgaaagcggatgaaaataatggatttggctcaaaaaatacagcgttctgcactcacgtctttcctgagactgcgtttatagcagtgacttcctaccagaaccacaagatcacgcaattaaagattgagaataatccctttgccaaaggatttcggggcagtgatgacatggagctgcacagaatgtcaagaatgcaaagtaaagaatatcccgtggtccccaggagcaccgtgaggcaaaaagtggcctccaaccacagtcctttcagcagcgagtctcgagctctctccacctcatccaatttggggtcccaataccagtgtgagaatggtgtttccggcccctcccaggacctcctgcctccacccaacccatacccactgccccaggagcatagccaaatttaccattgtaccaagaggaaagaggaagaatgttccaccacagaccatccctataagaagccctacatggagacatcacccagtgaagaagattccttctaccgctctagctatccacagcagcagggcctgggtgcctcctacaggacagagtcggcacagcggcaagcttgcatgtatgccagctctgcgccccccagcgagcctgtgcccagcctagaggacatcagctgcaacacgtggcc
aagcatgccttcctacagcagctgcaccgtcaccaccgtgcagcccatggacaggctaccctaccagcacttctccgctcacttcacctcggggcccctggtccctcggctggctggcatggccaaccatggctccccacagctgggagagggaatgttccagcaccagacctccgtggcccaccagcctgtggtcaggcagtgtgggcctcagactggcctgcagtcccctggcacccttcagccccctgagttcctctactctcatggcgtgccaaggactctatcccctcatcagtaccactctgtgcacggagttggcatggtgccagagtggagcgacaatagctaa,为野生型tbx5基因编码dna参考序列的第150位突变前碱基。
28.c.150c》g表示:与野生型tbx5基因编码dna的参考序列相比,突变基因的第150位点的碱基c突变为碱基g,具体编码dna的核苷酸序列为seq id no:3。
29.(4)野生型tbx5基因编码蛋白为:
30.megikvflherelwlkfhevgtemiitkagrrmfpsykvkvtglnpktkillmdivpaddhrykfadnkwsvtgkaepampgrlyvhpdspatgahwmrqlvsfqklkltnnhldpfghiilnsmhkyqprlhivkadenngfgskntafcthvfpetafiavtsyqnhkitqlkiennpfakgfrgsddmelhrmsrmqskeypvvprstvrqkvasnhspfssesralstssnlgsqyqcengvsgpsqdllpppnpyplpqehsqiyhctkrkeeecsttdhpykkpymetspseedsfyrssypqqqglgasyrtesaqrqacmyassappsepvpslediscntwpsmpsyssctvttvqpmdrlpyqhfsahftsgplvprlagmanhgspqlgegmfqhqtsvahqpvvrqcgpqtglqspgtlqppeflyshgvprtlsphqyhsvhgvgmvpewsdns。为野生型tbx5基因编码蛋白的第50位酪氨酸(tyr,y)。
31.p.tyr50ter表示:与野生型tbx5基因编码蛋白的氨基酸序列相比,突变基因编码蛋白的氨基酸第50位的酪氨酸(tyr,y)突变为终止密码子,具体氨基酸序列为seq id no:4。
32.(5)查询人群频率数据库发现tbx5基因c.150c》g杂合无义变异(tbx5:p.tyr50ter het)为罕见变异(千人基因组:无,esp6500:无,exac:无)。
33.该变异使第50位氨基酸处提前出现终止密码子,可能会使蛋白截短表达。
34.查询clinvar、hgmd数据库未发现该变异,该位置的缺失变异c.300del(p.lys99_tyr100inster)(与该变异导致的氨基酸改变相同)被上报者评定为致病突变(相关疾病:not provided,clinvar数据库);该位点下游的移码或无义变异多次被上报者评定为holt-oram综合征的致病突变(clinvar数据库),文献检索未发现该变异与疾病相关的报导。根据现有证据:该变异为罕见变异、该变异可能会使蛋白截短表达、该位置的其他变异以及下游的移码或无义变异多次被评定为致病突变、移码或无义变异为该基因主要致病类型,但缺乏家系连锁及功能学证据支持,所以该变异为holt-oram综合征的高度可疑致病突变(b级)。
35.实施例2-先天性心脏病突变基因的检测试剂盒
36.先天性心脏病突变基因的检测试剂盒,包括taq dna聚合酶、pcr缓冲液和引物等。具体引物如下:
37.上游引物(tbx5-e3f,seq id no:1):5'ctagtttccgcttccacgtt 3';
38.下游引物(tbx5-e3r,seq id no:2):5'gacagacgcctttagcacac 3';
39.长度:367bp。
40.利用本试剂盒筛选突变的致病基因tbx5的具体步骤为:提取待测者dna,然后使用经设计的引物组合(seq id no:1和seq id no:2)对tbx5基因进行扩增,得到pcr产物,使用1.5%的琼脂糖凝胶电泳检测pcr产物,选用1000bp marker作为参考,检测验证扩增产物为
预期的大小,最后对pcr产物进行测序。从ncbi(https://www.ncbi.nlm.nih.gov/)数据库获得参考序列和测序结果进行比对,判断待测者tbx5基因是否携带c.150c》g杂合错义变异,协助临床确诊holt-oram综合征患者。
41.实施例3-家系验证实验
42.本实施例采用家系连锁分析方法验证先天性心脏病突变基因的致病性。
43.具体地,选取一个家族性holt-oram综合征家系中的三代成员,该家系中的先证者(女,2岁9个月)临床被诊断为holt-oram综合征。
44.在先证者及其家属自愿签署知情同意书的前提下,寄送5-10ml全血样本,建立病历资料库,详细记录先证者病情、家系情况等资料。本研究已得到本单位伦理委员会批准。
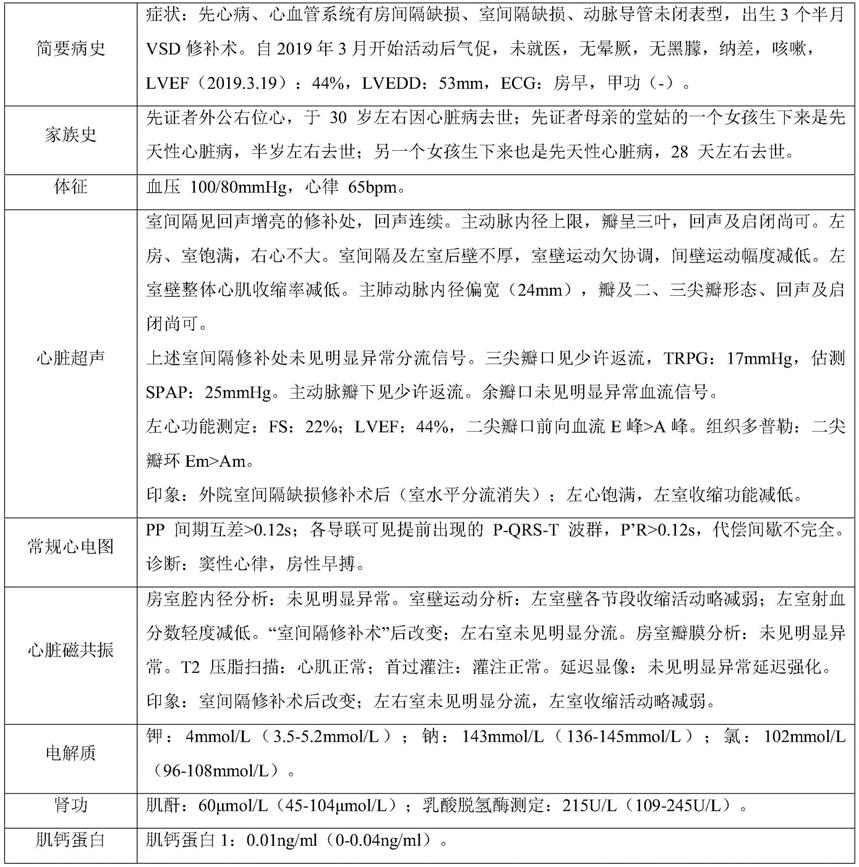
45.先证者临床概况描述:
46.表3先证者临床概况
[0047][0048]
采用实施例2提供的体外检测试剂盒对先证者及其家属的tbx5基因进行基因检测,结果如图1-图3所示,图1为实施例3的家系图;图2为实施例3中先证者、先证者母亲等人的sanger测序图;图3为实施例3家系中先证者父亲、先证者外婆等人的sanger测序图。
[0049]
如图1-图3所示,家系中临床诊断出holt-oram综合征的先证者、先证者母亲等人
均携带了突变的tbx5基因,而未患有holt-oram综合征的先证者父亲、先证者外婆均未携带突变的tbx5基因,由此验证可以说明突变的tbx5基因对holt-oram综合征的致病性。
[0050]
实施例4-针对家系外正常人的验证实验
[0051]
采用实施例2的holt-oram综合征检测试剂盒,对1150例家系外正常人进行tbx5基因检测,结果均未能检测到该突变。
[0052]
实施例5-针对家系外家族遗传性holt-oram综合征患者的验证实验
[0053]
在中国范围内,对患有holt-oram综合征、肥厚型心肌病、扩张型心肌病、长qt等单基因遗传病的患者中检测tbx5基因,患者总共2100例,每种疾病的患病人数不等,结果显示,除实施例3提供的家系外,仅在临床诊断患有holt-oram综合征的2例患者中检测到突变tbx5基因。
[0054]
本实验再次验证说明突变的tbx5致病基因的会导致holt-oram综合征,支持临床诊断。
[0055]
以上详细描述了本发明的较佳具体实施例。应当理解,本领域的普通技术人员无需创造性劳动就可以根据本发明的构思作出诸多修改和变化。因此,凡本技术领域中技术人员依本发明的构思在现有技术的基础上通过逻辑分析、推理或者有限的实验可以得到的技术方案,皆应在由权利要求书所确定的保护范围内。