1.本发明属于药物化学领域,具体涉及一种改进的酮咯酸的制备方法。
背景技术:
2.酮咯酸氨丁三醇是一种新型的可供注射的非甾体类强力止痛及中度抗炎解热药,其作用机理是通过抑制机体外周和中枢前列腺素(pg)的产生,从而降低外周和中枢中的pg,其主要成分酮咯酸的化学结构如下:
[0003][0004]
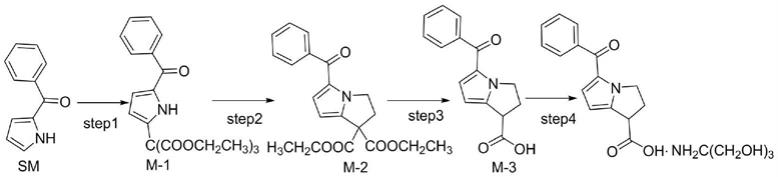
目前,酮咯酸氨丁三醇的合成多以2-苯甲酰吡咯为起始物料经过氧化缩合、合环、脱酸,最后成盐得到酮咯酸氨丁三醇,其反应路线如下:
[0005][0006]
在合成酮咯酸氨丁三醇的步骤中,酮咯酸的合成尤为重要。然而,现有文献资料公布的合成步骤中大多需要繁琐的后处理过程(中国药物化学杂志,1995,5(3):223-225;中国医药工业杂志,2001,32(5),202-203;),特别是浓缩的操作步骤难以避免,个别甚至采用柱层析的析法(us5082950,tetrahedron letters,2010,51(46):6000-6002)。此外,长时间的减压浓缩不仅延长了生产的周期,增加了生产的成本,而且长时间的加热还会增加中间体及酮咯酸产生杂质的几率,影响酮咯酸氨丁三醇最终的质量。然而,由于step1~step3反应中涉及多个中间体的合成,其每一步骤所用溶剂、试剂均可能对下一步产生干扰,特别是合成m-1的步骤中,大量锰离子和醋酸溶剂的使用会给后处理的过程带来分液萃取艰难及高沸点醋酸溶剂不易除去的难题,以往连续反应的愿望实则难以达到预期的目的。若醋酸溶剂富集至后续的合成反应中,则会给合环反应的发生带来较大的影响。再者,每个反应步骤完毕后大量存在的溶剂随之带来的大量三废问题也令人堪忧,而需要浓缩带来的高能耗和周期长等难题也给企业的生产成本带来了较大的挑战。
[0007]
专利cn101143865虽然对step2步骤合环反应和step3步骤水解脱羧反应的工艺进行了优化,尝试采用“一锅煮”的方法,但是没有指明中间体m-1的来源,可能涉及step1步骤氧化缩合反应存在大量金属离子和高沸点溶剂醋酸存在的难题,并且后续在step2步骤中的合环反应后处理过程中仍然采用了浓缩的操作。cn113603625和cn101575340报道,酮咯酸易于氧化。step1~step3的中间体稳定性不好,长时间的加热浓缩操作会影响酮咯酸的质量。因此,有必要对现有技术工艺进行改进。
[0008]
本发明人经过大胆的尝试和实验,反复摸索和研究,在专利cn113045471的基础上重点对step1中氧化缩合反应的合成条件进行优化,同时兼顾step2合环和step3水解脱羧
苯甲酰吡咯摩尔比约为0.05:1。
[0018]
优选的,在一具体实施方案中,本发明的一种改进的酮咯酸的制备方法,包含以下步骤:
[0019]
1)将2-苯甲酰吡咯、甲烷三羧酸三乙酯、醋酸锰二水合物(三价)、醋酸钠、醋酸酐加入到甲苯中,加热至反应完全;反应完后加入亚硫酸氢钠水溶液,分液,得到的有机相记为m-1溶液,直接进行下一步;
[0020]
2)加入m-1溶液,1,2-二氯乙烷,四丁基溴化铵,碳酸钾,适量水,加热至回流反应完全;加水分液,有机相用水洗涤两次,得到的有机相记为m-2溶液,直接进行下一步;
[0021]
3)加入m-2溶液,四丁基溴化铵,氢氧化钠水溶液,加热至至35~45℃反应完全;分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,干燥即可得到酮咯酸。
[0022]
其中,步骤1)所述亚硫酸氢钠与醋酸锰二水合物的摩尔比为(0.1~0.3):1.0。
[0023]
优选的,上述本发明的改进方法,步骤1)中所述甲苯的体积量为2-苯甲酰吡咯质量的为9-10倍。
[0024]
术语,所谓“体积量”是指每kg 2-苯甲酰吡咯所需溶剂的体积l量,即l/kg,或ml/g;比如甲苯溶剂的9~10倍体积量l(ml)是2-苯甲酰吡咯质量kg(g)的9~10倍。
[0025]
在一优选实施方案中,本发明的一种改进的酮咯酸的制备方法,包含:
[0026]
1)将2-苯甲酰吡咯、甲烷三羧酸三乙酯、醋酸锰二水合物(三价)、醋酸钠、醋酸酐加入到10倍体积量的甲苯中,加热至反应完全;加入0.1倍摩尔比的亚硫酸氢钠水溶液,分液,得到的有机相记为m-1溶液,直接进行下一步;
[0027]
2)加入m-1溶液,1,2-二氯乙烷,四丁基溴化铵,碳酸钾,适量水,加热至回流反应完全;加水分液,有机相用水洗涤两次,得到的有机相记为m-2溶液,直接进行下一步;
[0028]
3)加入m-2溶液,四丁基溴化铵,氢氧化钠水溶液,加热至至35~45℃反应完全;分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,干燥即可得到酮咯酸。
[0029]
在上述优选方案中,步骤1)中2-苯甲酰吡咯:甲烷三羧酸三乙酯:醋酸锰二水合物(三价):醋酸钠:醋酸酐的摩尔比为约1:1.5:1.5:2.0:7.5;
[0030]
在上述优选方案中,步骤2)中2-苯甲酰吡咯:1,2-二氯乙烷:碳酸钾:四丁基溴化铵=1:5.0~10.0:2.0:0.5:,优选为1:5.0~8.0:2.0:0.5,更优选为约1:6.0:2.0:0.5;
[0031]
在上述优选方案中,步骤3)中2-苯甲酰吡咯:四丁基溴化铵:氢氧化钠的摩尔比为1:0.05:(4.0~6.0),优选为约1:0.05:5.0。
[0032]
本文中“约”表示各步各种原料的投料量不一定是恰好是整数比,应当允许有偏差,通常在
±
0.1.
[0033]
依据本发明改进的制备方法得到酮咯酸,彻底地革除了现有工艺步骤中存在高能耗、高成本、周期长、污染大的繁琐操作步骤,解决了酮咯酸合成工业化大生产的难题。本发明的方法操作简单、成本低、污染少、纯度高、收率高。如下述实施例制得的酮咯酸,hplc检测纯度(面积归一法)大于或等于99.5%,单个最大杂质均在0.15%以下,三步总收率大于90%,适合于工业化大生产。得到的酮咯酸经过简单的脱色、成盐,得到的酮咯酸氨丁三醇hplc检测纯度大于或等于99.8%,单个最大杂质均在0.10%以下,符合药用的要求。
附图说明
[0034]
图1为实施例1获得的酮咯酸的hplc图谱。
[0035]
图2为实施例1获得的酮咯酸氨丁三醇的hplc图谱。
具体实施方式
[0036]
以下实施例仅是典型的,用于对本发明进行进一步说明和理解,但不以任何形式限制本发明的范围。
[0037]
液相色谱仪:岛津lc-2030c(3d plus)
[0038]
检测器:uv+dad
[0039]
色谱柱:inertsil ods-3(4.6*250mm,5μm)
[0040]
流动相:0.575%磷酸二氢铵溶液(3.0):四氢呋喃=70:30
[0041]
柱温:40℃
[0042]
流速:1.0ml/min
[0043]
波长:313nm
[0044]
进样量:10μl
[0045]
实施例1酮咯酸的制备
[0046]
合成路线:
[0047][0048]
将2-苯甲酰吡咯(sm)200g(1.17mol,1.0equiv.)、甲烷三羧酸三乙酯406.9g(1.75mol,1.5equiv.)、醋酸锰二水合物(三价)469.8g(1.75mol,1.5equiv.)、醋酸钠191.7g(2.34mol,2.0equiv.)、醋酸酐894.5g(8.76mol,7.5equiv.)加入到2000ml甲苯中,加热完全反应后,加入亚硫酸氢钠水溶液约1000ml(将54.7g亚硫酸氢钠(0.53mol,0.45equiv.)加入到1000ml水中进行溶解配置),分液,得到含有m-1中间体的有机相记为m-1溶液,直接进行下一步;
[0049]
将上述m-1溶液,1,2-二氯乙烷693.7g(7.01mol,6.0equiv.),四丁基溴化铵188.3g(0.58mol,0.5equiv.),碳酸钾322.9g(2.34mol,2.0equiv.),水322.9g,加热至回流反应完全;加水分液,有机相用水洗涤两次,得到含有m-2的中间体的有机相记为m-2溶液,直接进行下一步;
[0050]
将上述的m-2溶液,四丁基溴化铵18.8g(0.06mol,0.05equiv.),氢氧化钠水溶液约1168ml(将233.7g氢氧化钠(5.84mol,5.0equiv.)加入到1168ml水中进行溶解配置),加热至35~45℃反应完全;分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,真空干燥即可得到酮咯酸(m-3)约278.4g,三步总收率93.4%。经hplc检测,纯度99.72%,单个最大杂质0.13%。结果见表1和图1。将酮咯酸转化成酮咯酸氨丁三醇(参考cn101143865的制备方法),样品经hplc检测,纯度99.89%,单个最大杂质0.07%。结果见表2和图2。
[0051]
表1 所得酮咯酸的hplc检测结果
[0052]
峰号保留时间面积高度面积%分离度(usp)拖尾因子理论塔板数(usp)rrt18.6203185416980.13
‑‑
2.262810.420210.78538092890.025.7
‑‑
171960.526314.79531331550.019.2
‑‑
118800.721415.838158978850.062.11.3182460.772517.93270013310.034.2
‑‑
182360.874620.52124863339108232899.724.61.2187181.000742.74035531060.0132.31.1485072.083846.06952691320.024.41.2608132.245总计 249338531085924100.00
ꢀꢀꢀꢀ
[0053]
表2 所得酮咯酸氨丁三醇的hplc检测结果
[0054]
峰号保留时间面积高度面积%分离度(usp)拖尾因子理论塔板数(usp)rrt18.633159509010.07
‑‑
1.757040.420215.86443502530.0216.41.0221640.772320.55424086076104764399.899.11.2187331.000446.16861191420.0334.21.3422332.246总计 241124951048939100.00
ꢀꢀꢀꢀ
[0055]
实施例2
[0056]
将2-苯甲酰吡咯206g(1.20mol,1.0equiv.)、甲烷三羧酸三乙酯419.2g(1.81mol,1.5equiv.)、醋酸锰二水合物(三价)483.9g(1.81mol,1.5equiv.)、醋酸钠197.4g(2.41mol,2.0equiv.)、醋酸酐921.4g(9.03mol,7.5equiv.)加入到1850ml甲苯中,加热完全反应后,加入亚硫酸氢钠水溶液约1030ml(将41.3g亚硫酸氢钠(0.40mol,0.33equiv.)加入到1030ml水中进行溶解配置),分液,得到的有机相记为m-1溶液,直接进行下一步;
[0057]
将上述m-1溶液,1,2-二氯乙烷952.7g(9.63mol,8.0equiv.),四丁基氯化铵167.2g(0.60mol,0.5equiv.),碳酸钾332.6g(2.41mol,2.0equiv.),水332.6g,加热至回流反应完全;加水分液,有机相用水洗涤两次,得到的有机相记为m-2溶液,直接进行下一步;
[0058]
将上述的m-2溶液,四丁基氯化铵17.8g(0.06mol,0.05equiv.),氢氧化钠水溶液约963ml(将192.5g氢氧化钠(4.81mol,4.0equiv.)加入到963ml水中进行溶解配置),加热至35~45℃反应完全;分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,真空干燥即可得到酮咯酸约282.1g,三步总收率91.8%。经hplc检测,纯度99.67%,单个最大杂质0.12%。将制得的酮咯酸转化成酮咯酸氨丁三醇(参考cn101143865制备方法),样品经hplc检测,纯度99.84%,单个最大杂质0.06%。
[0059]
实施例3
[0060]
将2-苯甲酰吡咯183g(1.07mol,1.0equiv.)、甲烷三羧酸三乙酯372.4g(1.60mol,1.5equiv.)、醋酸锰二水合物(三价)429.9g(1.60mol,1.5equiv.)、醋酸钠175.4g(2.14mol,2.0equiv.)、醋酸酐818.5g(8.02mol,7.5equiv.)加入到1800ml甲苯中,加热完全反应后,加入亚硫酸氢钠水溶液约915ml(将50.1g亚硫酸氢钠(0.48mol,0.45equiv.)加入到915ml水中进行溶解配置),分液,得到的有机相记为m-1溶液,直接进行下一步;
[0061]
将上述m-1溶液,1,2-二氯乙烷528.9g(5.34mol,5.0equiv.),四丁基溴化铵
172.3g(0.53mol,0.5equiv.),碳酸钾295.5g(2.14mol,2.0equiv.),水295.5g,加热至回流反应完全;加水分液,有机相用水洗涤两次,得到的有机相记为m-2溶液,直接进行下一步;
[0062]
将上述的m-2溶液,四丁基氯化铵15.8g(0.05mol,0.05equiv.),氢氧化钾水溶液约1499ml(将299.8g氢氧化钾(5.34mol,5.0equiv.)加入到1499ml水中进行溶解配置),加热至35~45℃反应完全;分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,真空干燥即可得到酮咯酸约249.2g,三步总收率91.3%。经hplc检测,纯度99.64%,单个最大杂质0.14%。
[0063]
实施例4
[0064]
将2-苯甲酰吡咯215g(1.26mol,1.0equiv.)、甲烷三羧酸三乙酯437.5g(1.88mol,1.5equiv.)、醋酸锰二水合物(三价)505.1g(1.88mol,1.5equiv.)、醋酸钠206.1g(2.51mol,2.0equiv.)、醋酸酐961.6g(9.42mol,7.5equiv.)加入到2150ml甲苯中,加热完全反应后,加入亚硫酸氢钠水溶液约1075ml(将52.3g亚硫酸氢钠(0.50mol,0.40equiv.)加入到1075ml水中进行溶解配置),分液,得到的有机相记为m-1溶液,直接进行下一步;
[0065]
将上述m-1溶液,1,2-二氯乙烷745.7g(7.54mol,6.0equiv.),四丁基溴化铵202.4g(0.63mol,0.5equiv.),碳酸钾347.1g(2.51mol,2.0equiv.),水347.1g,加热至回流反应完全;加水分液,有机相用水洗涤两次,得到的有机相记为m-2溶液,直接进行下一步;
[0066]
将上述的m-2溶液,四丁基氯化铵18.6g(0.06mol,0.05equiv.),氢氧化钠水溶液约1507ml(将301.4g氢氧化钠(7.54mol,6.0equiv.)加入到1507ml水中进行溶解配置),加热至35~45℃反应完全;分液,水相用乙酸乙酯洗涤两次,降温,用盐酸调ph值1~2,过滤,滤饼用水洗涤,真空干燥即可得到酮咯酸约298.9g,三步总收率93.2%。经hplc检测,纯度99.69%,单个最大杂质0.12%。
[0067]
以上实施例仅是典型的,任何在本发明的精神实质下进行任何的简单修饰和变通都属于本发明的范围。