1.本发明属于高分子压电材料领域,公开了一种聚偏氟乙烯压电复合薄膜的制备方法。
背景技术:
2.随着全球对便携式、多功能化、轻量化自供电子设备的需求日益增加,对于拥有优异压电性能良好的材料被用作自供电电子设备的重点原材料已经成为众多学者研究的热点。高分子压电材料因为其声阻抗低、柔顺性好和容易与水以及人体等轻质负载匹配的优点,这是传统的陶瓷和石英以及无机压电材料所没有的。然而,聚偏氟乙烯(pvdf)的结构灵活、压电系数高、生物兼容性好、容易加工等优点,是具有代表性的压电材料。pvdf作为一种半结晶型高分子,共有多达五种晶型,其中β晶为全反式构象,晶胞中含有极性锯齿,因此β-pvdf具有最好的压电性能。但是纯pvdf压电系数偏低,难以满足传感器和能量收集器件等自供电电子设备
37.对该类材料的使用要求。因此提升pvdf的β相相对含量对于其应用至压电传感器具有重要的意义,但制备具有高β相含量的pvdf薄膜仍是一个待解决的问题。
3.为了提高pvdf的压电性能,选择加入无机填料,其中包括石墨烯、碳纳米管(cnts)、粘土、蒙脱土、沸石等,其诱导β-pvdf形成的机理为:填料中正电荷/负电荷粒子和pvdf中的cf2/ch2基团之间的相互作用。β相的诱导程度在很大程度上取决于纳米材料的性质(如类型、形状和尺寸)、表面化学能和分散状态。其中cnts具有特有的纳米一维管状结构,由多个石墨烯片绕着某一中心旋转堆叠形成中空的柱状结构,这样独特的一维空间结构赋予cnts优异的性能,具体表现在填充到聚合物材料中,可明显增强其机械性能、电性能和其他物理方面的性能。但cnts具有团聚效应,且cnts本身具有强作用力,这导致在聚合物集体中容易发生团聚现象;又由于cnts在基体材料中无序排列,使cnts在聚合物基体中均匀分散是有效发挥填充材料cnts性能的关键。
4.对于cnts的功能化改性中包括共价和非共价功能化改性。非共价官能化主要是基于范德华力、氢键、静电力和π-π共轭相互作用而进行的一些超分子络合。虽然其改性过程温和,但表面活性剂改性的cnts由于与基体之间的相互作用力差,只能使聚合物基体的力学性能得到缓和的提高,即聚合材料性能的时效性差。因此选用共价官能化改性cnts。共价官能化就是在碳纳米管表面形成官能团,官能团参与聚合反应,共价官能化不仅能够有效的改善碳纳米管在溶剂中的分散性,且由于其改善了界面粘附性,因此只需要添加微量就可以得到很好的效果。所以,发展高效,低能耗和绿色的聚合物修饰cnts的新技术,进而能够使它的化学反应更加实用且绿色,是cnts功能化研究发展的重要目的和方向。
技术实现要素:
5.针对上述问题,本发明基于碳纳米管接枝聚离子液体提出一种聚偏氟乙烯压电复合薄膜的制备方法。
6.本发明通过以下技术方案实现:
7.一种聚偏氟乙烯压电复合薄膜的制备方法,包括以下步骤:
8.(1)将初始的碳纳米管用浓硫酸与浓硝酸的混合溶液进行酸化处理,得到羧基化碳纳米管;再使用二氯亚砜处理得到酰氯化碳纳米管;而后使用甲苯、乙二胺及4-二甲氨基吡啶处理得到氨基化碳纳米管;最后使用无水三乙胺、2-溴代异丁酰溴得到溴化碳纳米管。
9.(2)通过乙烯基类与咪唑类烯烃反应合成离子液体单体备用。
10.(3)通过表面引发氧化-还原聚合将步骤(1)得到的溴化碳纳米管表面接枝步骤(2)得到碳纳米管接枝聚离子液体,简称cnts@pil。
11.(4)将步骤(3)中得到的cnts@pil添加到pvdf基体材料中搅拌均匀,除去溶剂后熔融压模,得pvdf薄膜。
12.(5)将步骤(4)中得到的pvdf薄膜极化封装,得聚偏氟乙烯压电复合薄膜。
13.进一步的,步骤(1)中所述的原始碳纳米管为长度0.5-2μm,内径为5-15nm,纯度>95%。
14.进一步的,步骤(2)中所述的乙烯基类为4-氯甲基苯乙烯、甲基苯乙烯、乙烯基苯乙烯中的任意一种。
15.进一步的,步骤(2)中所述的咪唑类烯烃为二甲基咪唑或乙基咪唑。
16.进一步的,步骤(2)中所述的离子液体单体为氯离子型离子液体、六氟磷酸根型离子液体、四氟硼酸根型离子液体中的任意一种或多种。
17.进一步的,步骤(3)中使用引发剂为过硫酸铵、过硫酸钾或过硫酸钠中的一种,引发剂用量为离子液体单体质量的1%-2%,聚合温度为45-65℃,聚合时间为8-16h,氧化-还原聚合反应接枝在有机溶剂二甲基亚砜中进行,且引发剂为aibn,其中还使用了配体三(2-二甲氨基乙基)胺,催化剂溴化铜,所述离子液体单体与溴化碳管的质量比为10:1-30:1。
18.进一步的,步骤(4)中所添加的cnts@pil为pvdf质量的1-10%。
19.进一步的,步骤(5)中的封装工艺为首先在pvdf薄膜上用导电银胶粘上铜片,在60-80℃烘箱中固化1-6h,再旋涂聚二甲基硅氧烷(pdms)旋涂液(pdms旋涂液制备方法为将pdms:固化剂以质量比10:1搅拌5min,真空除去气泡。旋涂工艺为:旋涂转速1000r/min,旋涂时间30-60s),旋涂完毕后在60-80℃下固化1-6h,然后将涂覆pvdf的ito玻璃反压在上述旋涂pdms后的pvdf薄膜上,在60-80℃下干燥(一般干燥1-4h),室温下固化(一般固化24h左右),在ito玻璃涂覆pvdf的一面以同样方式粘上铜片并以裁剪好的pp塑料封装。
20.本发明与现有技术相比具有如下有益效果:本发明采用共价改性的方法,对cnts表面修饰改性接枝聚离子液体,因其与pvdf之间强的π-π相互作用及氢键作用,能有效提高pvdf的极性晶相的生成,制备得到高效的压电薄膜。
附图说明
21.图1为实例1中氯离子型液体单体的核磁谱图。
22.图2为实例2中六氟磷酸根离子型液体单体的核磁谱图。
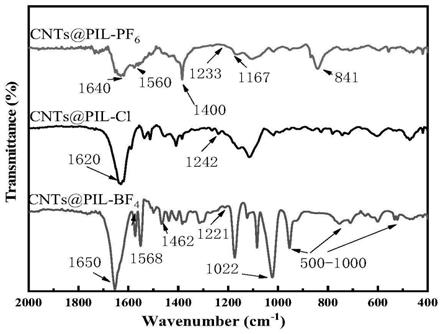
23.图3为实例1、实例2及实例3中cnts接枝离子液体cnts@pil-bf4、cnts@pil-pf6、cnts@pil-cl的红外光谱图。
24.图4为实例1、实例2及实例3中pvdf及pvdf-cnts@pil复合膜的介电常数(左)以及其对应的介电损耗对比图。
25.图5为实例1、实例2及实例3中pvdf及pvdf-cnts@pil复合膜的压电常数。
26.图6为不同cnts添加量在pvdf薄膜中的红外光谱图,从上至下依次为pvdf/10wt%cnts、pvdf/5wt%cnts、pvdf/1wt%cnts、pvd。
具体实施方式
27.本发明不局限于下列具体实施方式,本领域一般技术人员根据本发明公开的内容,可以采用其他多种具体实施方式实施本发明的,或者凡是采用本发明的设计结构和思路,做简单变化或更改的,都落入本发明的保护范围。需要说明的是,在不冲突的情况下,本发明中的实施例及实施例中的特征可以相互组合。
28.本发明下面结合实施例作进一步详述:
29.一、本发明所使用的试剂均为市购,检测仪器有:
30.红外光谱(ftir):使用傅立叶变换红外光谱仪(型号avatar 370)(热电尼高力仪器公司,美国),使用制备完成的样品进行红外测试并分析,ftir通过记录4000和500cm-1之间32次扫描得到,分辨率为2cm-1。根据beer-lambert,ftir测试结果其吸收峰面积计算β晶型的含量。β晶含量计算公式如下:
[0031][0032]
式中:aa为761cm-1
处α的最大吸收强度;a
β
为836cm-1
相的最大吸收强度;ka=6.0
×
104cm2/mol、k
β
=7.4
×
104cm2/mol,为相应的吸收系数。
[0033]
waxd衍射测试:运用x射线在晶体中衍射,可以得到晶体结构、晶胞参数等。还有助于观察,分析晶相。设置测试速度6
°
/min,2θ角扫射范围0-60℃,选择cu-kα射线进行测量。
[0034]
dsc测试:首先选用较高浓度的氮气作为保护气,将不同水平因素的样本置于机器内。以10℃/min,从50℃升温至200℃,恒温3min,观察并记录第一次升温曲线。设置降温速率为10℃/min降至50℃,恒温3min,目的是消除热历史,观察并记录其降温结晶曲线。再进行第二次升温,实验参数同第一次升温一致。
[0035]
sem测试:扫描电镜可以对样品在一定的放大倍数下的表面、断面形貌进行观察,可以得到薄膜改性前后晶粒尺寸的变化。测试表面形貌之前将pvdf薄膜剪成尺寸为1cm
×
1cm,然后用导电胶固定在圆柱模具上,对膜进行表面喷金、观察表面形貌。对于薄膜断面形貌的测试,先将尺寸为1cm
×
2cm的薄膜放置于液氮中让其脆断,再次将薄膜固定,进行喷金,测试。
[0036]
压电性能测试:材料压电性能主要通过压电系数进行表征,将样品进行裁剪成1cm
×
1cm的样品,先将薄膜用铝箔包裹,先用硅油热极化,置于10kv的高压极化场下,电场强度为4000v/m,温度设置为90℃,持续时间1h,待极化后的样品冷却到室温后,再将包裹好的薄膜置于d
33
仪器上进行测试。
[0037]
介电性能测试:先将待测薄膜裁剪成1cm
×
1cm的样品,将薄膜两面用导电胶带粘贴好,而后使用agilent 429a型精密阻抗分析仪进行样品的自由电容、介电损耗的测试,测试的频率范围为:25~106hz。薄膜样品的介电常数可以通过一下公式得到:
[0038]
[0039]
其中εr:介电常数;cp:特定频率下的电容(f/m);d:薄膜的厚度;ε0:真空介电常数(8.854
×
10-12
f/m)s:薄膜两侧导电胶的面积(cm2)。
[0040]
二、pvdf压电薄膜制备方案的具体步骤
[0041]
实施例1
[0042]
一种pvdf压电薄膜制备的方案,由以下方法制备:
[0043]
步骤1,羧基化碳纳米管制备:1-5gcnts加入到50-200ml的混酸中(体积比:浓硫酸:浓硝酸=3:1),在30-80℃下超声4-6h,而后洗涤抽滤至滤液呈中性,常温干燥保存。
[0044]
步骤2,酰氯化碳纳米管的制备:将步骤1制备的羧基化碳纳米管均匀分散在30-100ml甲苯中,滴加30-100ml二氯亚砜,60-100℃下回流反应6-24h。洗涤后常温干燥保存。
[0045]
步骤3,氨基化碳纳米管的制备:将步骤2的酰氯化碳纳米管,2-5g的4-二甲氨基吡啶,100-200ml甲苯以及50-100ml乙二胺于100℃下回流6-24h。洗涤后常温干燥保存。
[0046]
步骤4:溴化碳纳米管的制备:将步骤3的氨基化碳纳米管分散在乙腈溶液中加入1-3ml无水乙胺和1-3ml的2-溴代异丁酰胺的乙腈溶液3.00-5ml。洗涤后常温干燥保存。
[0047]
步骤5:将1份氯甲基苯乙烯和1份二甲基咪唑于乙腈溶液中,在室温下反应24-72h,最终得到的为氯离子型离子液体单体。将氯离子型离子液体单体溶解在饱和的六氟磷酸盐溶液中,室温搅拌3-12h,干燥后室温保存的到六氟磷酸根型离子液体单体。将氯离子型离子液体单体溶解在饱和的四氟硼酸盐溶液中,室温搅拌3-12h,干燥后室温保存的到四氟硼酸根型离子液体单体。
[0048]
步骤6:将步骤4的碳纳米管与步骤5得到的氯离子型离子液体单体在二甲基亚砜溶剂中,添加配体三(2-二甲氨基乙基)胺,引发剂aibn,催化剂溴化铜在80-100℃下反应6-24h。离心洗涤干燥,得碳纳米管接枝氯离子型聚离子液体(cnts@pil-cl)粒子,室温保存。本步骤中的配体三(2-二甲氨基乙基)胺可以调节过渡金属的氧化还原性能,并以自身的立体效应供给催化剂以适当的选择性,还可以改善催化剂在反应混合物中的溶解性能,此外,还可以选用二联吡啶作为配体。
[0049]
步骤7:将步骤6得到的1重量份cnts@pil-cl粒子加入到95重量份pvdf中,搅拌均匀,而后减压蒸馏将溶剂除去,得到的产物在10mpa,180℃下熔融模压3min,得pvdf薄膜。
[0050]
步骤8:将步骤7得到的pvdf薄膜进行极化后封装。首先在pvdf薄膜上用导电银胶粘上铜片,在60-80℃烘箱中固化1-6h,再旋涂聚二甲基硅氧烷(pdms)旋涂液(pdms旋涂液制备方法为将pdms:固化剂以质量比10:1搅拌5min,真空除去气泡。旋涂工艺为:旋涂转速1000r/min,旋涂时间30-60s),旋涂完毕后在60-80℃下固化1-6h,然后将涂覆pvdf的ito玻璃反压在上述旋涂pdms后的pvdf薄膜上,在60-80℃下干燥(一般干燥1-4h),室温下固化(一般固化24h左右),在ito玻璃涂覆pvdf的一面以同样方式粘上铜片并以裁剪好的pp塑料封装。
[0051]
实施例2
[0052]
一种pvdf压电薄膜制备的方案,由以下方法制备:
[0053]
步骤1:1-5gcnts加入到50-200ml的混酸中(浓硫酸:浓硝酸=3:1),在30-80℃下超声4-6h,而后洗涤抽滤至滤液呈中性,常温干燥保存。
[0054]
步骤2:将步骤1的cnts均匀分散在30-100ml甲苯中,滴加30-100ml二氯亚砜,60-100℃下回流反应6-24h。洗涤后常温干燥保存。
[0055]
步骤3:将步骤2的cnts,2-5g的4-二甲氨基吡啶,100-200ml甲苯以及50-100ml乙二胺于100℃下回流6-24h。洗涤后常温干燥保存。
[0056]
步骤4:将步骤3的cnts的乙腈溶液中加入1-3ml无水乙胺和1-3ml的2-溴代异丁酰胺的乙腈溶液3.00-5ml。洗涤后常温干燥保存。
[0057]
步骤5:将1份氯甲基苯乙烯和1份二甲基咪唑于乙腈溶液中,在室温下反应24-72h,最终得到的为氯离子型离子液体单体。将氯离子型离子液体单体溶解在饱和的六氟磷酸盐溶液中,室温搅拌3-12h,干燥后室温保存的到六氟磷酸根型离子液体单体。将氯离子型离子液体单体溶解在饱和的四氟硼酸盐溶液中,室温搅拌3-12h,干燥后室温保存的到四氟硼酸根型离子液体单体。
[0058]
步骤6:将步骤4的cnts分别与步骤5得到的六氟磷酸根型离子液体单体在二甲基亚砜溶剂中,添加配体三(2-二甲氨基乙基)胺,引发剂aibn,催化剂溴化铜在80-100℃下反应6-24h。离心洗涤干燥,得碳纳米管接枝六氟磷酸根离子型聚离子液体(cnts@pil-pf6)粒子,室温保存。
[0059]
步骤7:将步骤6得到的1重量份碳纳米管接枝六氟磷酸根离子型聚离子液体(cnts@pil-pf6)粒子加入到95重量份pvdf中,在适量dmf溶剂中搅拌均匀,而后减压蒸馏将溶剂除去,得到的产物在10mpa,180℃下熔融模压3min。
[0060]
步骤8:将步骤7得到的pvdf薄膜进行极化后封装。首先在pvdf薄膜上用导电银胶粘上铜片,在60-80℃烘箱中固化1-6h,再旋涂聚二甲基硅氧烷(pdms)旋涂液(pdms旋涂液制备方法为将pdms:固化剂以质量比10:1搅拌5min,真空除去气泡。旋涂工艺为:旋涂转速1000r/min,旋涂时间30-60s),旋涂完毕后在60-80℃下固化1-6h,然后将涂覆pvdf的ito玻璃反压在上述旋涂pdms后的pvdf薄膜上,在60-80℃下干燥(一般干燥1-4h),室温下固化(一般固化24h左右),在ito玻璃涂覆pvdf的一面以同样方式粘上铜片并以裁剪好的pp塑料封装。
[0061]
实施例3
[0062]
一种pvdf压电薄膜制备的方案,由以下方法制备:
[0063]
步骤1:1-5gcnts加入到50-200ml的混酸中(浓硫酸:浓硝酸=3:1),在30-80℃下超声4-6h,而后洗涤抽滤至滤液呈中性。常温干燥保存。
[0064]
步骤2:将步骤1的cnts均匀分散在30-100ml甲苯中,滴加30-100ml二氯亚砜,60-100℃下回流反应6-24h。洗涤后常温干燥保存。
[0065]
步骤3:将步骤2的cnts,2-5g的4-二甲氨基吡啶,100-200ml甲苯以及50-100ml乙二胺于100℃下回流6-24h。洗涤后常温干燥保存。
[0066]
步骤4:将步骤3的cnts的乙腈溶液中加入1-3ml无水乙胺和1-3ml的2-溴代异丁酰胺的乙腈溶液3.00-5ml。洗涤后常温干燥保存。
[0067]
步骤5:将1份氯甲基苯乙烯和1份二甲基咪唑于乙腈溶液中,在室温下反应24-72h,最终得到的为氯离子型离子液体。将氯离子型离子液体溶解在饱和的六氟磷酸盐溶液中,室温搅拌3-12h,干燥后室温保存的到六氟磷酸根型离子液体。将氯离子型离子液体溶解在饱和的四氟硼酸盐溶液中,室温搅拌3-12h,干燥后室温保存的到四氟硼酸根型离子液体。
[0068]
步骤6:将步骤4的cnts分别与步骤5得到的四氟硼酸根型离子液体在二甲基亚砜
溶剂中,添加配体三(2-二甲氨基乙基)胺,引发剂aibn,催化剂溴化铜在80-100℃下反应6-24h。离心洗涤干燥,得碳纳米管接枝四氟硼酸根离子型聚离子液体(cnts@pil-bf4)粒子,室温保存。
[0069]
步骤7:将步骤6得到的1份碳纳米管接枝四氟硼酸根离子型聚离子液体(cnts@pil-bf4)粒子加入到95份pvdf中,在适量dmf溶剂中搅拌均匀,而后减压蒸馏将溶剂除去,得到的产物在10mpa,180℃下熔融模压3min。
[0070]
步骤8:将步骤7得到的pvdf薄膜进行极化后封装。首先在pvdf薄膜上用导电银胶粘上铜片,在60-80℃烘箱中固化1-6h,再旋涂聚二甲基硅氧烷(pdms)旋涂液(pdms旋涂液制备方法为将pdms:固化剂以质量比10:1搅拌5min,真空除去气泡。旋涂工艺为:旋涂转速1000r/min,旋涂时间30-60s),旋涂完毕后在60-80℃下固化1-6h,然后将涂覆pvdf的ito玻璃反压在上述旋涂pdms后的pvdf薄膜上,在60-80℃下干燥(一般干燥1-4h),室温下固化(一般固化24h左右),在ito玻璃涂覆pvdf的一面以同样方式粘上铜片并以裁剪好的pp塑料封装。
[0071]
实施例4
[0072]
一种pvdf压电薄膜制备的方案,由以下方法制备:
[0073]
步骤1:将原始的1份cnts加入到95份pvdf中,在适量dmf溶剂中搅拌均匀,而后减压蒸馏将溶剂除去,得到的产物在10mpa,180℃下熔融模压3min。
[0074]
步骤2:将步骤1得到的pvdf薄膜进行极化后封装。首先在pvdf薄膜上用导电银胶粘上铜片,在60-80℃烘箱中固化1-6h,再旋涂聚二甲基硅氧烷(pdms)旋涂液(pdms旋涂液制备方法为将pdms:固化剂以质量比10:1搅拌5min,真空除去气泡。旋涂工艺为:旋涂转速1000r/min,旋涂时间30-60s),旋涂完毕后在60-80℃下固化1-6h,然后将涂覆pvdf的ito玻璃反压在上述旋涂pdms后的pvdf薄膜上,在60-80℃下干燥(一般干燥1-4h),室温下固化(一般固化24h左右),在ito玻璃涂覆pvdf的一面以同样方式粘上铜片并以裁剪好的pp塑料封装。
[0075]
三、表征与性能测试
[0076]
图1为实例1中氯离子型液体单体的核磁谱图。通过分析核磁氢谱与单体结构的相符性来判断此单体是否成功合成,在低场7.6ppm的峰为咪唑环上的两个氢质子,高场2.7、3.9ppm处为单一尖峰对应于甲基的氢质子,是咪唑环上的甲基,δ=5.4ppm处为亚甲基中的氢质子,7.2-7.3ppm处的峰为苯环上的两个氢质子,δ=6.7ppm处为乙烯基与苯环相连的碳上的氢质子,峰面积相等的两组峰(δ约5.3、5.9处)为乙烯基末端碳上的两个氢质子,各峰面积为3:3:1:1:2:1:1:1:1:1这符合il-cl中各氢质子数量比,证明成功合成il-cl单体。
[0077]
图2为实例2中六氟磷酸根型液体单体的核磁谱图。在低场7.6-7.7ppm峰为咪唑环上的两个氢质子,高场2.7、3.9ppm处为单一尖峰对应于甲基的氢质子,7.3和7.5ppm的峰为苯环上的两个氢质子,h场6.7ppm为盼多重峰(对应一个氢质子)是由乙烯基和苯环基团耦合裂分产生的,5.3和5.9ppm的峰为乙烯基末端碳上的两个氢质子,各峰面积比为3:3:1:1:2:1:1:1:1:1,这符合il-pf6中各氢质子数量比,证明成功合成il-pf6单体。
[0078]
图3为实例1、实例2及实例3中cnts接枝离子液体cnts@pil-bf4、cnts@pil-pf6、cnts@pil-cl的红外光谱图。从图中可以看到在1640、1620cm-1
和1650cm-1
处均为cnts中不饱和c=c键的特征峰,1560cm-1
、1568cm-1
c处均为咪唑环骨架的振动吸收峰,1233cm-1
、
1242cm-1
和1221cm-1
处均为咪唑环上的c-n的振动吸收峰,而cnts@pil-bf4在1022cm-1
处出现b-f键强吸收峰,cnts@pil-pf6在1167cm-1
、1400cm-1
分别出现了c-n和c-h的特征峰,说明乙二胺成功接枝,841cm-1
处的峰对应于p-f特征峰,上述特征峰的变化充分说明atrp成功制备了cnts@pil-bf4,cnts@pil-pf6。cnts@pil-cl由于其吸水性较大,不太稳定,其红外谱图无明显的其他特征峰。
[0079]
图4为实例1、实例2及实例3中pvdf及pvdf-cnts@pil复合膜的介电常数(左)以及其对应的介电损耗对比图(右)。pvdf-cnts@pil复合薄膜在电压为1v条件下,介电常数与频率的关系,及频率在1000hz下其介电损耗。由公式2.2计算出pvdf的介电常数有266左右,加入cnts后其介电常数增加至380左右,当加入改性后的cnts@pil,其介电常数有更加明显的增加,得到其介电常数可以达到413-532左右。表明离子液体对于碳纳米管的修饰使其在界面交换耦合效应和mws极化机理有不同的促进作用。而不同阴离子的离子液体,其对于cnts的氢键作用大不相同,bf
4-及pf
6-相比与cl-具有更多的f-,而f-的强电负性能与cnts上的-oh有强的氢键作用,其对于提高cnts的作用性及分散性更加有效,因此m-pvdf-cnts@pil-pf6具有更高的介电常数,高达532。可以看出,添加了cnts的薄膜相比于纯pvdf,其介电损耗有明显增加,这是因为cnts作为一种电学性能优良的碳材料,易形成导电通路,从而使得漏电流增多,介电损耗增加。但cnts经过离子液体改性后,cnts和il的之间的π-π或cation-π相互作用,il可以对cnts团簇进行剥离,提高其分散性,使得导电网络形成困难,漏电流减小,因此在相同的cnts含量添加下,pvdf-cnts@pil-pf6的介电损耗仅有0.01075。
[0080]
图5为实例1、实例2及实例3中pvdf及pvdf-cnts@pil复合膜的压电常数图。纯m-pvdf和m-pvdf-cnts@pil压电薄膜的压电常数d
33
数值。从图中可以看出,与纯m-pvdf薄膜相比,m-pvdf-cnts@pil薄膜的压电常数有明显的增大,综合ftir、xrd以及dsc的数据分析显示,这主要是由于m-pvdf-cnts@pil复合薄膜中β相的相对含量提高了。相比与纯m-pvdf的d
33
值为3pc/n,pvdf-cnts@pil-pf6的d
33
值增大到21pc/n。原因主要是pvdf-cnts@pil-pf6中的β相的相对含量高,且其具备较高的介电常数和较低的介电损耗,其极化能力增强,两方面综合作用,使得其压电常数d
33
值最高,压电性能最佳。
[0081]
图6为前期筛选碳纳米管添加量,分别选用1、3、5重量份碳纳米管添加到pvdf中。从红外谱图中可以明显看出添加碳纳米管后的pvdf薄膜的极性β相的相对含量有所增加。
[0082]
以上显示和描述了本发明的基本原理和主要特征和本发明的优点。本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明精神和范围的前提下,本发明还会有各种变化和改进,这些变化和改进都落入要求保护的本发明范围内。本发明要求保护范围由所附的权利要求书及其等效物界定。
[0083]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,根据本发明的技术方案及其构思加以等同替换或改变,都应涵盖在本发明的保护范围之内。