1.本发明属于阻燃材料领域,具体涉及一种磷氮芳香族阻燃剂及其制备方法和应用。
背景技术:
2.环氧树脂固化后形成三维交联网状结构,种类繁多,力学强度高、化学性质稳定,应用领域广泛,例如建材、电子电器、航空航天、汽车、包装等。环氧树脂具有优异的综合性能,然而其极限氧指数《22%属于易燃材料,燃烧过程中产生大量浓烟和熔融滴落物,对人类的生存环境有极大的安全威胁,易燃性限制了环氧树脂的应用,因此有必要对环氧树脂进行阻燃改性。卤系阻燃剂在热裂解或者燃烧时,会释放大量的烟尘和有毒腐蚀性气体,基于环保和健康考虑,目前使用量减少,发展受限;磷系阻燃剂具有成碳性,可减少可燃物的生成,还有抗氧化作用,能有效提高阻燃效率,但现有磷系阻燃剂在环氧树脂中存在易析出,不易分散的技术难题。单独使用二氨基二苯基甲烷(ddm)与环氧树脂的固化产物室温使用期短,固化产物耐热性有限;使用间苯二胺(mpd)与环氧树脂的固化产物熔点高室温使用不方便且工艺较为复杂。
技术实现要素:
3.本发明的目的是要解决现有磷系阻燃剂在环氧树脂中存在易析出,不易分散的技术难题,而提供磷氮阻燃剂化合物及其制备方法和利用其制备阻燃环氧树脂的方法。
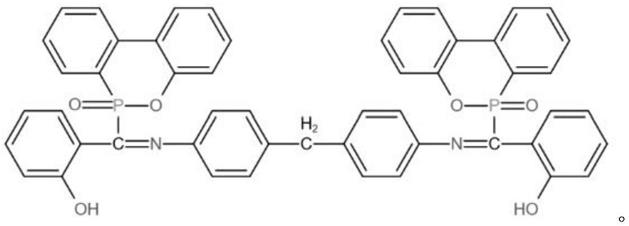
4.磷氮阻燃剂化合物,它的结构式为:
[0005][0006]
磷氮阻燃剂化合物的制备方法,具体是按以下步骤完成的:先由二氨基二苯基甲烷和水杨醛在溶剂中脱水缩合制成中间体亚胺sd,再由中间体亚胺sd和9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物在无水乙醇中磷氢加成反应得到磷氮阻燃剂化合物。
[0007]
利用磷氮阻燃剂化合物制备阻燃环氧树脂的方法,具体是按以下步骤完成的:先将环氧树脂加热至90~120℃,再加入磷氮阻燃剂化合物,并混合均匀,然后冷却至温度为70~90℃,再加入固化剂ddm-mpd,混合均匀后真空脱泡,再依次进行浇注模具、两级固化和自然冷却,得到阻燃环氧树脂。
[0008]
本发明原理及优点:
[0009]
一、本发明合成的磷氮阻燃剂化合物比传统卤素阻燃剂环保,无污染,无腐蚀性气
体产生,比现有含磷阻燃剂的制备条件更简单,反应条件易控制,反应过程无危险产物生成,阻燃效率更高;
[0010]
二、本发明以磷氮阻燃剂化合物与环氧树脂混合,再加入固化剂,解决了现有磷系阻燃剂在环氧树脂中存在易析出,不易分散的技术问题;
[0011]
三、本发明的阻燃环氧树脂可根据阻燃要求的不同,通过改变阻燃剂添加量,制备得到不同情况下使用的阻燃环氧树脂,有利于节省成本;
[0012]
四、本发明合成的阻燃环氧树脂过程设备简单,步骤简单,无高温高压,制备过程安全,原料无有害物质,工业化生产价值高。
[0013]
五、本发明合成的阻燃环氧树脂与原树脂相比,表现出了优异的阻燃性能,垂直燃烧固化物中磷含量低于0.6%。
[0014]
六、本发明合成的磷氮阻燃剂化合物及阻燃环氧树脂具有一定的韧性和耐热性,可以用于建材、汽车、航空、包装等领域。
附图说明
[0015]
图1为实施例1得到的磷氮阻燃剂化合物的红外光谱图;
[0016]
图2为实施例1得到的磷氮阻燃剂化合物的核磁共振氢谱图;
[0017]
图3为图2中化学位移6cm-1
~8.5cm-1
范围的放大图;
[0018]
图4为条状待测试件的结构示意图;
[0019]
图5为哑铃状待测试件的结构示意图。
具体实施方式
[0020]
具体实施方式一:本实施方式是磷氮阻燃剂化合物,它的结构式为:
[0021][0022]
具体实施方式二:本实施方式是磷氮阻燃剂化合物的制备方法,具体是按以下步骤完成的:先由二氨基二苯基甲烷和水杨醛在溶剂中脱水缩合制成中间体亚胺sd,再由中间体亚胺sd和9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物在无水乙醇中磷氢加成反应得到磷氮阻燃剂化合物。
[0023]
具体实施方式三:本实施方式与具体实施方式二的不同点是:所述溶剂为无水乙醇。其他与具体实施方式二相同。
[0024]
具体实施方式四:本实施方式与具体实施方式二或三之一不同点是:所述脱水缩合的操作过程如下:在温度为60~80℃下搅拌反应5h~7h。其他与具体实施方式二或三相同。
[0025]
先将本实施方式得到的脱水缩合产物进行自然冷却,再真空过滤,然后用无水乙醇洗涤2次,最后在温度为50~70℃下干燥12h,得到中间体亚胺sd,封袋备用。
[0026]
具体实施方式五:本实施方式与具体实施方式二至四之一不同点是:所述二氨基二苯基甲烷与水杨醛的摩尔比为(0.4~0.7):1;所述二氨基二苯基甲烷的物质的量与溶剂的体积比为1mol:(4700~5300)ml。其他与具体实施方式二至四相同。
[0027]
具体实施方式六:本实施方式与具体实施方式二至五之一不同点是:所述磷氢加成反应的操作过程如下:在温度为60~80℃下搅拌反应2h~5h。其他与具体实施方式二至五相同。
[0028]
先将本实施方式得到的磷氢加成反应产物进行自然冷却,再减压过滤,然后用无水乙醇洗涤2次,最后在温度为50~70℃下干燥12h,得到磷氮阻燃剂化合物,封袋备用。
[0029]
具体实施方式七:本实施方式与具体实施方式二至六之一不同点是:所述中间体亚胺sd与9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物的摩尔比为(0.4~0.7):1;所述9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物与无水乙醇的体积比为1mol:(2700~3300)ml。其他与具体实施方式二至六相同。
[0030]
具体实施方式八:本实施方式是利用磷氮阻燃剂化合物制备阻燃环氧树脂的方法,具体是按以下步骤完成的:先将环氧树脂加热至90~120℃,再加入磷氮阻燃剂化合物,并混合均匀,然后冷却至温度为70~90℃,再加入固化剂ddm-mpd,混合均匀后真空脱泡,再依次进行浇注模具、两级固化和自然冷却,得到阻燃环氧树脂。
[0031]
本实施方式所述固化剂ddm-mpd为具有低熔点的共熔混合状态的芳香胺类固化剂。
[0032]
具体实施方式九:本实施方式与具体实施方式二的不同点是:所述固化剂ddm-mpd由间苯二胺和二氨基二苯基甲烷加热熔融混合而成,且所述间苯二胺和二氨基二苯基甲烷的质量比(1.4~1.7):1;所述环氧树脂为e51双酚a型缩水甘油醚类环氧树脂;所述两级固化具体操作过程如下:先在温度为60~90℃下固化2h,然后在温度为140~160℃下固化2h。其他与具体实施方式九相同。
[0033]
本实施方式固化剂ddm-mpd的具体制备过程如下:将间苯二胺(mpd)和二氨基二苯基甲烷(ddm)在烧杯中混合,然后加热熔融混合均匀,得到固化剂ddm-mpd。
[0034]
具体实施方式十:本实施方式与具体实施方式九或十之一不同点是:所述阻燃环氧树脂中磷氮阻燃剂化合物的质量分数为5%~20%;所述环氧树脂的环氧基与固化剂ddm-mpd的活泼氢当量比(0.9~1.2):1。其他与具体实施方式九或十相同。
[0035]
本发明内容不仅限于上述各实施方式的内容,其中一个或几个具体实施方式的组合同样也可以实现发明的目的。
[0036]
采用下述试验验证本发明效果:
[0037]
实施例1:磷氮阻燃剂化合物的制备方法,具体是按以下步骤完成的:
[0038]
一、脱水缩合:将无水乙醇加入配有磁力搅拌器和回流冷凝管的三口圆底烧瓶中,再加入二氨基二苯基甲烷和水杨醛,在温度为70℃下搅拌反应6h,得到的脱水缩合产物,先自然冷却至室温,再真空过滤,然后用无水乙醇洗涤2次,最后放入真空干燥箱内,在温度为60℃下干燥12h,得到金黄色固体,即为中间体亚胺sd;所述二氨基二苯基甲烷与水杨醛的摩尔比为0.5:1;所述二氨基二苯基甲烷的物质的量与溶剂的体积比为1mol:5000ml;
[0039]
二、磷氢加成:将无水乙醇加入配有磁力搅拌器和回流冷凝管的三口圆底烧瓶中,再加入中间体亚胺sd和9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物,在温度为70℃下搅拌反
应3h,得到的磷氢加成反应产物,先自然冷却至室温,再减压过滤,然后用无水乙醇洗涤2次,最后放入真空干燥箱内,在温度为60℃下干燥12h,得到淡黄色固体,即为磷氮阻燃剂化合物(sd-dopo);所述中间体亚胺sd与9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物的摩尔比为0.5:1;所述9,10-二氢-9-氧杂-10-磷杂菲-10-氧化物与无水乙醇的体积比为1mol:3000ml。
[0040]
分别采用核磁共振氢谱仪和傅里叶红外光谱仪对实施例1得到的磷氮阻燃剂化合物进行测试分析,如图1、图2和图3,图1为实施例1得到的磷氮阻燃剂化合物的红外光谱图;图2为实施例1得到的磷氮阻燃剂化合物的核磁共振氢谱图,图3为图2中化学位移6cm-1
~8.5cm-1
范围的放大图;通过图2和图3可以看出,在h
8-15
范围内所示为dopo各类质子吸收峰,δ=9.51ppm和δ=9.38ppm处的两个特征峰为sd-dopo上两个酚羟基的质子峰,δ=9.51ppm和δ=9.38ppm处出现两个-oh信号;通过图1可知:1274cm-1
处出现芳香族c-n单键的伸缩振动峰,红外结果证明dopo的加成反应使得c=n双键被打开发生加成反应生成c-n单键,在图谱中也没有发现p-h键的红外特征吸收峰。结果中还发现1612cm-1
处为p-ph键,1212cm-1
处为p=o键的特征吸收峰,927cm-1
处强吸收峰为p-o-ph结构,以上分析表明sd和dopo之间发生加成反应,成功的合成了磷氮阻燃剂化合物(sd-dopo),其结构式为:
[0041]
实施例2:利用磷氮阻燃剂化合物制备阻燃环氧树脂的方法,具体是按以下步骤完成的:将间苯二胺(mpd)和二氨基二苯基甲烷(ddm)在烧杯中混合,然后加热熔融混合均匀,得到固化剂ddm-mpd;所述固化剂ddm-mpd由间苯二胺和二氨基二苯基甲烷加热熔融混合而成,且所述间苯二胺和二氨基二苯基甲烷的质量比1.5:1;将环氧树脂加热至110℃,再加入实施例1得到的磷氮阻燃剂化合物和丙酮(丙酮作为溶剂使实施例1得到的磷氮阻燃剂化合物与环氧树脂混合均匀),并混合均匀,待丙酮完全去除后,冷却至温度为80℃,再加入固化剂ddm-mpd,混合均匀后真空脱泡,然后注入预热模具中,转移至烘箱中,在温度为70℃下固化2h,然后在温度为150℃下固化2h,自然冷却至室温,得到阻燃环氧树脂;所述环氧树脂的环氧基与固化剂ddm-mpd的活泼氢当量比1:1;所述预热模具的预热温度为70℃;所述环氧树脂为e51双酚a型缩水甘油醚类环氧树脂;所述阻燃环氧树脂中磷氮阻燃剂化合物的质量分数为5%。
[0042]
实施例3:本实施例与实施例2的不同点是:所述阻燃环氧树脂中磷氮阻燃剂化合物的质量分数为10%。其他与实施例2相同。
[0043]
实施例4:本实施例与实施例2的不同点是:所述阻燃环氧树脂中磷氮阻燃剂化合物的质量分数为15%。其他与实施例2相同。
[0044]
实施例5:本实施例与实施例2的不同点是:所述阻燃环氧树脂中磷氮阻燃剂化合物的质量分数为20%。其他与实施例2相同。
[0045]
按照ul94astm d635-77测定e51双酚a型缩水甘油醚类环氧树脂和实施例2至5得
到的阻燃环氧树脂的垂直燃烧ul-94,如表1所示。将e51双酚a型缩水甘油醚类环氧树脂和实施例2至5得到的阻燃环氧树脂制成条状待测试件,条状待测试件的长度为130mm,宽度为6.5mm,厚度为3mm,然后按照gb/t2406-2009测定条状待测试件的极限氧指数loi;将e51双酚a型缩水甘油醚类环氧树脂和实施例2至5得到的阻燃环氧树脂制成哑铃状待测试件,哑铃状待测试件的长度为130mm,厚度为4mm,哑铃状待测试件两端的宽度为20mm
±
0.5mm,哑铃状待测试件中间的宽度为10mm
±
0.2mm,且宽度为10mm
±
0.2mm中间段的长度为50mm
±
0.5mm,哑铃状待测试件的中间段与两端采用弧形过渡,单侧弧形过渡段的长度为32.5mm;按照gb/t1040-2006测定哑铃状待测试件的拉伸性能;测试结果如表2所示。
[0046]
表1
[0047][0048]
表2
[0049][0050]
通过表1和表2,由e51双酚a型缩水甘油醚类环氧树脂与实施例2至5得到的阻燃环氧树脂对比可知,通过添加本发明制备的磷氮阻燃剂化合物,能够显著提高环氧树脂的阻燃性能,实施例5得到的阻燃环氧树脂极限氧指数loi达到34.2%;且通过实施例2至5得到的阻燃环氧树脂之间对比可知,通过改变磷氮阻燃剂化合物添加量,制备得到不同情况下使用的阻燃环氧树脂;根据力学性能测试结果可知,该阻燃环氧树脂的断裂强度能够保持在20mpa以上。