1.本发明涉及合成润滑油技术领域,更具体地说,是涉及一种超高粘度指数酯类基础油及其制备方法。
背景技术:
2.润滑油主要是用于降低设备负荷、提高工作效率、减少设备磨损的油状液体。除润滑作用外,它还具有冷却、防锈、防腐、清洁、密封、缓冲、功率传送及电气绝缘等作用,因而被广泛应用于运输业、工业、农业、军事、医药、航空航天及日常生活等领域。一个国家润滑油行业的发展状况直接反映了其经济发达程度、机械制造水平、设备控制状况、装置工作效率及环境保护意识。近年来,随着国民经济的高速发展与汽车保有量的持续增长,我国润滑油消费总量已位于世界前列。据统计,2017~2019年我国润滑油基础油的表观消费量分别为674、617、648万吨,供需比相应为92%、96%、97%,表明润滑油基础油仍处于供不应求的状态。另一方面,对高性能润滑油需求量的日益增大不断倒逼我国润滑油产业的结构升级,促使润滑油行业从数量增长模式向质量发展模式转变,表明高性能润滑油具有广阔的市场应用前景。润滑油通常由基础油与添加剂调和而成,其中基础油约占润滑油质量的80~99%,对润滑油性能起主导作用,因此高品质基础油是生产高性能润滑油的先决条件。由此可见,对高品质基础油的研究开发及生产具有重要的现实意义。
3.矿物基础油作为目前占据市场份额最多的基础油,其综合使用性能一般,在自然环境中难以生物降解,且原料不可再生。生物基础油虽然具有润滑性与生物降解性好、原料可再生等优点,但其合成原料中的非理想组分较多,产品氧化安定性、热稳定性及低温流动性较差,目前尚难得到广泛应用。作为一种高端的合成基础油,酯类基础油具有润滑性、粘温性能及低温流动性能出众,热稳定性与氧化安定性较好,蒸发损失小,溶解性好,易生物降解且无毒等优点,可兼顾高性能、多功能性及环保性。此外,酯类基础油的分子构型设计灵活,理化性能调控方便,在合成超高粘度指数基础油时具有显著优势。据统计,2017年我国对酯类基础油的需求量约为3.93万吨,而产量为2.55万吨;预计到2023年,我国对酯类基础油的需求量将超过6万吨。因此,用于生产高性能的环保型超高粘度指数润滑油的酯类基础油具有广阔的市场前景。
4.在目前报道的酯类基础油中,蔡国星与魏梅莹等人合成了具有“哑铃结构”的酯类基础油,他们先利用新戊基多元醇与二元羧酸进行酯化反应,合成出多羟基寡聚物,再以一元羧酸为封端剂,酯化未反应的羟基。虽然引入二元羧酸可通过“桥联作用”增加酯分子的碳链长度,提高其粘度指数,但该方法存在以下弊端:(1)分子构型设计不合理,该酯类油的粘度指数难以达到170以上;(2)油品中重组分含量较多,长期储存易产生白色沉淀;(3)对甲苯磺酸催化剂会导致产品的硫含量过高,反应副产物较多;(4)以甲苯为携水剂,有损操作人员的身体健康,易造成环境污染并增大能耗;(5)采用碱洗精制法,污水排放量大,产品收率低。美国专利us6774093b2公开了一种以直链脂肪酸、支链脂肪酸及季戊四醇为原料合成聚季戊四醇混合酸酯的方法。该方法借助季戊四醇分子间的醚化反应延长分子的主碳链
长度与支链数,精制后的聚季戊四醇混合酸酯的运动粘度(100℃)、粘度指数及倾点分别为19.2mm2·
s-1
、97、-32℃。显而易见,该基础油的粘温性能并不理想。虽然复酯具有很高的粘度指数,但其分子结构设计不灵活,β氢原子较多。
5.综上所述,上述文献报道的酯类基础油及其合成方法均存在一定缺陷,而高性能酯类基础油的绿色合成已呈不可逆转趋势,故应从分子构型设计、绿色催化剂制备、绿色合成及精制工艺四方面加以改善。
技术实现要素:
6.有鉴于此,本发明的目的在于提供一种超高粘度指数酯类基础油及其制备方法,本发明提供的超高粘度指数酯类基础油的综合性能较好且具有极佳的储存稳定性。
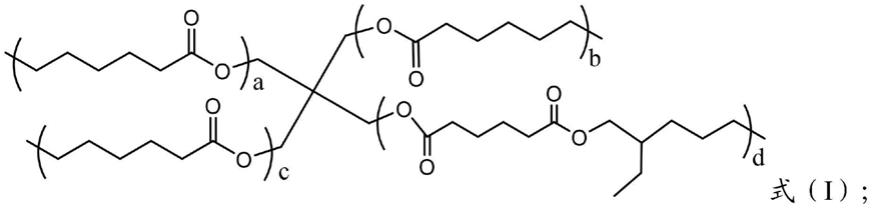
7.本发明提供了一种超高粘度指数酯类基础油,具有式(i)~(iii)所示的结构:
[0008][0009][0010][0011]
其中,a、b、c、d分别为封端结构的基团数量,a+b+c+d=单分子新戊基多元醇的羟基数。
[0012]
本发明还提供了一种上述技术方案所述的具有式(i)所示结构的超高粘度指数酯类基础油的制备方法,包括以下步骤:
[0013]
a)在氮气保护下先将二元羧酸、一元醇和锡锆复合氧化物催化剂混合,进行第一步反应;再加入一元羧酸、新戊基多元醇和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物;
[0014]
b)将步骤a)得到的反应混合物中的锡锆复合氧化物催化剂及非理想组分分离,得
到超高粘度指数的酯类基础油。
[0015]
本发明还提供了一种上述技术方案所述的具有式(ii)所示结构的超高粘度指数酯类基础油的制备方法,包括以下步骤:
[0016]
a)在氮气保护下先将二元羧酸、一元醇和锡锆复合氧化物催化剂混合,进行第一步反应;再加入新戊基多元醇和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物;或者,在氮气保护下先将混合二元羧酸、新戊基多元醇和锡锆复合氧化物催化剂混合,进行第一步反应;再加入一元醇和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物;
[0017]
b)将步骤a)得到的反应混合物中的锡锆复合氧化物催化剂及非理想组分分离,得到超高粘度指数的酯类基础油。
[0018]
本发明还提供了一种上述技术方案所述的具有式(iii)所示结构的超高粘度指数酯类基础油的制备方法,包括以下步骤:
[0019]
a)在氮气保护下先将适量的二元羧酸、一元醇和锡锆复合氧化物催化剂混合,进行第一步反应;再加入新戊基多元醇、剩余量的二元羧酸和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物;或者,在氮气保护下先将适宜量的二元羧酸、新戊基多元醇和锡锆复合氧化物催化剂混合,进行第一步反应;再加入一元醇、剩余量的二元羧酸和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物;
[0020]
b)将步骤a)得到的反应混合物中的锡锆复合氧化物催化剂及非理想组分分离,得到超高粘度指数的酯类基础油。
[0021]
优选的,步骤a)中所述保护氮气的通入量≤3ml
·
min-1
·
g-1
。
[0022]
优选的,步骤a)中所述新戊基多元醇选自新戊基二醇、三羟甲基丙烷、季戊四醇、双三羟甲基丙烷和双季戊四醇中的一种或多种;所述二元羧酸选自c4~c
12
中的一种或多种;所述一元醇选自c6~c
10
中的一种或多种;所述一元羧酸选自c6~c
18
中的一种或多种。
[0023]
优选的,步骤a)中所述锡锆复合氧化物催化剂中锡与锆的摩尔比为(6~10):1;
[0024]
所述锡锆复合氧化物催化剂的加量为反应物料总质量的1.0wt.%~3.0wt.%。
[0025]
优选的,步骤a)中所述第一步反应的温度为140℃~170℃,反应时间为1h~2.5h;所述第二步反应的具体条件为:先在160℃~170℃下反应0.5h~2h,再升温至170℃~180℃,反应3h以上。
[0026]
优选的,步骤b)中所述油剂分离的方式为真空抽滤;所述真空抽滤的温度为110℃~150℃。
[0027]
优选的,所述步骤b)还包括:
[0028]
将分离后得到的双酯型新戊基多元醇酯粗产品采用两级分子蒸馏进行精制,最终得到精制产品。
[0029]
本发明提供了一种超高粘度指数酯类基础油及其制备方法;所述超高粘度指数的酯类基础油为双酯型新戊基多元醇酯,具有三大亚类,其分子结构分别如式(i)~(ⅲ)所示,式(i)~(ⅲ)分别为c类、b类及a类双酯型新戊基多元醇酯的分子结构式;其中a、b、c、d分别为封端结构的基团数量,a+b+c+d=单分子新戊基多元醇的羟基数,例如当以季戊四醇为新戊基多元醇原料时,a+b+c+d=4;需要强调的是,新戊基多元醇、二元羧酸、一元羧酸及一元醇原料并不局限于上述原料。本发明涉及一类超高粘度指数酯类基础油的构型设计与绿色合成方法。鉴于对环保型高性能润滑油基础油需求量的逐渐增大,本发明从分子构型、
酯化催化剂、合成工艺及精制方法四方面入手,设计并合成了a、b、c三类双酯型新戊基多元醇酯。该双酯型新戊基多元醇酯可用作环保型高性能润滑油的基础油。与文献报道的高粘度指数酯类基础油不同,本发明设计的双酯型新戊基多元醇酯的分子构型更为合理,具有更高的粘度指数、更低的倾点。同时,与传统生产方式采用浓硫酸、对甲苯磺酸及氯化亚锡等催化剂不同,本发明所用的锡锆复合氧化物催化剂具有催化活性高、残留低且易于油剂分离等优点,可避免s、n、p及卤族元素的残留,减少反应副产物、产品乳化及设备腐蚀,并简化精制工艺;与传统生产方式采用甲苯、二甲苯等携水剂不同,本发明以氮气为携水剂,可避免有毒有机溶剂对操作人员身体健康的损害,减少环境污染,并降低能耗;与传统生产方式采用碱洗精制不同,本发明采用分子蒸馏精制法,可有效脱除水、有机酸、半酯及低沸点酯等非理想组分,不仅增大了精制深度,减少了环境污染,避免了产品乳化,还可实现资源的充分利用。
附图说明
[0030]
图1为本发明实施例提供的c类双酯型季戊四醇酯的
13
c-nmr谱(a)及dept 135
°
谱(b);
[0031]
图2为本发明实施例提供的c类双酯型季戊四醇酯的
13
c-nmr谱的化学位移预测;
[0032]
图3为本发明实施例提供的c类双酯型季戊四醇酯的1h-nmr谱;
[0033]
图4为本发明实施例提供的c类双酯型季戊四醇酯的1h-nmr谱的化学位移预测。
具体实施方式
[0034]
下面将结合本发明实施例,对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0035]
本发明提供了一种超高粘度指数酯类基础油,具有式(i)~(iii)所示的结构:
[0036][0037]
[0038][0039]
其中,a、b、c、d分别为封端结构的基团数量,a+b+c+d=单分子新戊基多元醇的羟基数。
[0040]
本发明设计并合成出一类超高粘度指数的酯类基础油,具有三大亚类,其分子结构分别如上述式(i)~(ⅲ)所示;式(i)~(ⅲ)分别为c类、b类及a类双酯型新戊基多元醇酯的分子结构式,其中a、b、c、d分别为封端结构的基团数量,a+b+c+d=单分子新戊基多元醇的羟基数,例如当以季戊四醇为新戊基多元醇原料时,a+b+c+d=4。需要强调的是,新戊基多元醇、二元羧酸、一元羧酸及一元醇原料并不局限于上述原料(新戊基多元醇的分子结构差异较大,例如三羟甲基丙烷与季戊四醇的分子结构式分别如下式所示。上述分子通式是以季戊四醇为例说明,而合成时实施例1~2则是以三羟甲基丙烷为新戊基多元醇原料。事实上,三羟甲基丙烷、季戊四醇、新戊基二醇、双三羟甲基丙烷及双季戊四醇均可作为新戊基多元醇原料)。
[0041]
三羟甲基丙烷的分子结构式:
[0042]
季戊四醇的分子结构式:
[0043]
本发明设计并合成的超高粘度指数酯类基础油以新戊基多元醇及一元醇为醇原料,以二元羧酸和一元羧酸为酸原料,其中每一个二元羧酸分子分别与新戊基多元醇和一元醇分子相连,从而形成“类双酯结构”。
[0044]
本发明设计并合成的超高粘度指数酯类基础油具有较低的倾点、极高的粘度指数、较好的储存稳定性,进而能够解决现有桥联型酯类基础油粘度指数难以达到很高且易产生白色沉淀的问题。
[0045]
本发明还提供了一种上述技术方案所述的超高粘度指数酯类基础油的制备方法,包括以下步骤:
[0046]
a)合成反应在氮气保护下进行,对于c类双酯型新戊基多元醇酯,先将二元羧酸、一元醇(摩尔比为1:1)和锡锆复合氧化物催化剂混合,进行第一步反应;再加入一元羧酸、新戊基多元醇和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物。对于b类双酯型
新戊基多元醇酯,先将二元羧酸、一元醇(摩尔比为1:1)和锡锆复合氧化物催化剂混合,进行第一步反应;再加入新戊基多元醇和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物;亦或者,先将混合二元羧酸、新戊基多元醇和锡锆复合氧化物催化剂混合,进行第一步反应,再加入一元醇和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物。对于a类双酯型新戊基多元醇酯,先将二元羧酸、一元醇(摩尔比为1:1)和锡锆复合氧化物催化剂混合,进行第一步反应;再加入新戊基多元醇、剩余量的二元羧酸和锡锆复合氧化物催化剂进行第二次反应,得到反应混合物;亦或者,先将适宜量的二元羧酸、新戊基多元醇和锡锆复合氧化物催化剂混合,进行第一步反应,再加入一元醇、剩余量的二元羧酸(摩尔比为1:1)和锡锆复合氧化物催化剂进行第二步反应,得到反应混合物。
[0047]
b)将步骤a)得到的反应混合物中的锡锆复合氧化物催化剂及非理想组分进行分离,得到超高粘度指数的酯类基础油。
[0048]
由上可知,本发明提供的制备方法涉及两步反应,简单地讲,可分为先形成“半酯结构”再形成“双酯结构”,亦或者是先形成“多羟基寡聚物”再利用一元醇“封头”。对于c类及b类双酯型新戊基多元醇酯,一元醇与二元羧酸的摩尔比为1:1,其区别在于a类、b类酯全部由一元醇“封头”,而c类酯由一元羧酸和一元醇共同“封头”。若将b类酯看作是双酯型新戊基多元醇酯的基本构型,则c类双酯型新戊基多元醇酯可看作是b类双酯型新戊基多元醇酯与新戊基多元醇酯的复合酯,a类双酯型新戊基多元醇酯可看作是b类双酯型新戊基多元醇酯与桥联型新戊基多元醇酯的复合酯。
[0049]
与传统生产方式采用甲苯、二甲苯或环己烷等有毒的有机携水剂不同,本发明以氮气为携水剂,可避免有毒有机溶剂对操作人员身体的损害,减少环境污染,防止产品氧化,并降低能耗。在本发明中,所述保护氮气的通入量优选≤3ml
·
min-1
·
g-1
,更优选为2ml
·
min-1
·
g-1
。
[0050]
在本发明中,所述新戊基多元醇优选选自新戊基二醇、三羟甲基丙烷、季戊四醇、双三羟甲基丙烷和双季戊四醇中的一种或多种,更优选为季戊四醇和三羟甲基丙烷。在本发明优选的实施例中,所述新戊基多元醇为季戊四醇和三羟甲基丙烷。本发明对所述新戊基多元醇的来源没有特殊限制,采用本领域技术人员熟知的上述新戊基二醇、三羟甲基丙烷、季戊四醇、双三羟甲基丙烷和双季戊四醇的市售商品或自制品均可。
[0051]
在本发明中,所述二元羧酸优选选自c4~c
12
中的一种或多种,更优选为己二酸、戊二酸、壬二酸和癸二酸。本发明对所述二元羧酸的来源没有特殊限制,采用本领域技术人员熟知的上述己二酸、戊二酸、壬二酸和癸二酸的市售商品或自制品均可。
[0052]
与传统生产方式采用浓硫酸、对甲苯磺酸及氯化亚锡等催化剂不同,本发明所用的锡锆复合氧化物催化剂具有催化活性高、残留低且易于油剂分离等优点,可避免s、n、p及卤族元素的残留,减少反应副产物、产品乳化及设备腐蚀,并简化精制工艺。在本发明中,所述锡锆复合氧化物催化剂优选采用水热法合成;所述水热法的合成温度优选为150℃~190℃,水热时间优选为12h~18h;得到的催化剂产品的粒径优选为20目~40目。在本发明中,所述锡锆复合氧化物催化剂不含s、n、p及卤族元素,制备过程不使用模板剂与表面活性剂。
[0053]
在本发明中,所述锡锆复合氧化物催化剂中锡与锆的摩尔比优选为(6~10):1。在本发明中,所述锡锆复合氧化物催化剂在第一步反应中的加量优选为反应物总质量的1.0wt.%~3.0wt.%,更优选为1.5wt.%。
[0054]
本发明对所述混合的方式没有特殊限制,采用本领域技术人员熟知的机械搅拌或人工搅拌的技术方案均可;在本发明优选的实施例中,所述混合的方式采用机械搅拌;所述机械搅拌的转速优选为600rpm~1200rpm,更优选为800rpm。
[0055]
在本发明中,所述第一步反应的温度优选为140℃~170℃,更优选为160℃;所述第一步反应的时间优选为1h~2.5h,更优选为1.5h。
[0056]
本发明在整个反应过程中(包括第一步反应和第二步反应),通入氮气进行保护;所述保护氮气的通入量优选≤3ml
·
min-1
·
g-1
,更优选为2ml
·
min-1
·
g-1
;同时,本发明在优选的实施例中,进行机械搅拌,所述机械搅拌的转速优选为600rpm~1200rpm,更优选为800rpm。
[0057]
在本发明中,所述一元醇优选选自c6~c10中的一种或多种,更优选为2-乙基己醇、正庚醇中的一种或多种,更更优选为2-乙基己醇。本发明对所述一元醇的来源没有特殊限制,采用本领域技术人员熟知的上述2-乙基己醇、正庚醇的市售商品或自制品均可。
[0058]
在本发明中,所述一元羧酸优选选自c6~c18中的一种或多种,更优选自正己酸、正庚酸和2-乙基己酸中的一种或多种,更优选为正庚酸。本发明对所述一元羧酸的来源没有特殊限制,采用本领域技术人员熟知的上述正己酸、正庚酸和2-乙基己酸的市售商品或自制品均可。
[0059]
在本发明优选的实施例中,所述第二步反应进一步加入锡锆复合氧化物催化剂,加量优选为第二步反应所加反应物总质量的1.0wt.%~3.0wt.%,更优选为1.50wt.%。
[0060]
在本发明中,所述第二次反应的条件优选具体为:
[0061]
先在160℃~170℃下反应0.5h~2h,再升温至170℃~180℃,反应3h以上;
[0062]
更优选为:
[0063]
先在170℃下反应1h,再升温至180℃,反应6~7h。
[0064]
得到所述反应混合物后,本发明将得到的反应混合物中的锡锆复合氧化物催化剂进行分离,得到超高粘度指数的酯类基础油粗产品。
[0065]
在本发明中,所述催化剂分离的方式优选为真空抽滤;为了在提高油剂分离效率的同时防止产品氧化,所述真空抽滤的温度优选为110℃~150℃。
[0066]
在本发明中,当催化剂与酯类润滑油基础油分离时,真空度非稳定状态,视真空泵的性能、新旧、密封状况及抽滤过程而变,真空表示数范围为-0.1mpa~0mpa。当真空泵较新、性能较好或抽滤初期,真空表示数接近为-0.1mpa,当真空泵较旧、性能一般、密封较差或抽滤中期,真空表示数则高于-0.1mpa,在抽滤中后期直至结束,真空表示数逐渐变为0mpa。
[0067]
本发明优选还包括:
[0068]
将分离后得到的双酯型新戊基多元醇酯粗产品采用两级分子蒸馏进行精制,得到精制产品。
[0069]
与采用传统的碱洗精制方式不同,本发明采用分子蒸馏精制法,可有效脱除水、有机酸、半酯及低沸点酯等非理想组分,不仅增大了精制深度、减少了环境污染、避免了产品乳化,还可实现资源的充分利用。
[0070]
本发明以两级分子蒸馏精制代替传统的碱洗精制;所述两级分子蒸馏的第一级分子蒸馏主要脱除有机酸、水、半酯及低沸点酯,第二级分子蒸馏脱除残余的微量有机酸,使
其酸值达到行业标准要求;即精制后分离出水、有机酸、半酯及低沸点酯等非理想组分,最终得到精制产品。在本发明中,所述两级分子蒸馏条件具体优选为:
[0071]
蒸发器温度为150℃~180℃,内冷器温度≤15℃,绝对压力≤3pa;半酯、低沸点酯的分离量可根据凝胶色谱测定的轻组分含量确定;若第一级分子蒸馏所得产品的酸值>0.05mgkoh/g,可进行第二级分子蒸馏精制,蒸出少许轻组分即可。
[0072]
本发明提供的制备方法可得到综合性能较好的超高粘度指数酯类基础油,并且属于绿色合成方法,能够解决传统酯类基础油合成及精制过程中污染物排放量大、产品硫含量超标、反应副产物多等问题。
[0073]
本发明提供了一种超高粘度指数酯类基础油及制备方法;所述超高粘度指数的酯类基础油为双酯型新戊基多元醇酯,具有三大亚类,其分子结构分别如式(i)~(ⅲ)所示,式(i)~(ⅲ)分别为c类、b类及a类双酯型新戊基多元醇酯的分子结构式;其中a、b、c、d分别为封端结构的基团数量,a+b+c+d=单分子新戊基多元醇的羟基数,例如当以季戊四醇为新戊基多元醇原料时,a+b+c+d=4;需要强调的是,新戊基多元醇、二元羧酸、一元羧酸及一元醇原料并不局限于上述原料。本发明涉及一类超高粘度指数酯类基础油的构型设计与绿色合成方法。鉴于对环保型高性能润滑油基础油需求量的逐渐增大,本发明从分子构型、酯化催化剂、合成工艺及精制方法四方面入手,设计并合成了a、b、c三类双酯型新戊基多元醇酯。该双酯型新戊基多元醇酯可用作环保型高性能润滑油的基础油。与文献报道的高粘度指数酯类基础油不同,本发明设计的双酯型新戊基多元醇酯的分子构型更为合理,具有更高的粘度指数、更低的倾点。同时,与传统生产方式采用浓硫酸、对甲苯磺酸及氯化亚锡等催化剂不同,本发明所用的锡锆复合氧化物催化剂具有催化活性高、残留低且易于油剂分离等特点,可避免s、n、p及卤族元素的残留,减少反应副产物、产品乳化及设备腐蚀,并简化精制工艺;与传统生产方式采用甲苯、二甲苯等携水剂不同,本发明以氮气为携水剂,可避免有毒有机溶剂对操作人员身体健康的损害,减少环境污染,并降低能耗;与传统生产方式采用碱洗精制不同,本发明采用分子蒸馏精制法,可有效脱除水、有机酸、半酯及低沸点酯等非理想组分,不仅增大了精制深度,减少了环境污染,避免了产品乳化,还可实现资源的充分利用。
[0074]
为进一步说明本发明,下面通过以下实施例进行详细说明。本发明以下实施例中所用的催化剂为锡锆复合氧化物,其制备所用试剂如表1所示。
[0075]
表1锡锆复合氧化物制备所用试剂
[0076]
名称规格厂商氯化锡(ⅱ)二水合物ar,98%macklin氯氧化锆八水合物ar,99%aladdin去离子水二次蒸馏自制氨水(25wt.%)ar天津市富宇精细化工有限公司无水乙醇ar北京化工厂硝酸银ar上海第一试剂厂
[0077]
所述锡锆复合氧化物的制备方法如下:(1)将0.2mol/l的zrocl2溶液、sncl2·
2h2o粉末及去离子水按一定比例配制成体积约为80ml的混合盐溶液,在快速搅拌下加入适量氨水,调节其ph至目标值;(2)在室温条件下搅拌1h后,将催化剂母液倒入容量为100ml、带有
聚四氟乙烯内衬的水热釜中,并用氮气吹扫液面上方的空气,按照一定温度与时间在油浴锅中进行水热处理;(3)过滤反应后的产物,用去离子水和无水乙醇交替洗涤沉淀物,直至滤液用硝酸银溶液检测不到cl-为止;(4)将过滤得到的固体置于真空干燥箱中,在60℃条件下干燥12h即可得到锡锆复合氧化物。
[0078]
所述锡锆复合氧化物催化剂的适宜锡锆摩尔比为(6~10):1,ph为8.5~9.5,水热温度为160℃~180℃,水热时间为16h~20h。
[0079]
所用的其他原料均为市售商品。
[0080]
实施例1
[0081]
a类双酯型新戊基多元醇酯粗产品的合成:将三羟甲基丙烷、己二酸及2-乙基己醇按2:5:4的摩尔比分两步加入反应体系,第一步加入摩尔比为2:1的三羟甲基丙烷与己二酸,先形成“桥联结构”;第二步再加入摩尔比为4:4的己二酸及2-乙基己醇,形成“双酯结构”。第一、二步反应的反应温度均为170℃,反应时间分别为1h与4h,催化剂加量均为反应物料总质量的1%,氮气流量为2ml
·
min-1
·
g-1
,搅拌速度为800rpm。待反应结束后,在真空条件下通过抽滤实现超高粘度指数酯类基础油与锡锆复合氧化物催化剂的分离,得到a类双酯型新戊基多元醇酯的粗产品。
[0082]
在上述合成条件下制得的a类双酯型新戊基多元醇酯粗产品在40℃及100℃时的运动粘度、粘度指数、色度及倾点分别为1246.08mm2/s、140.79mm2/s、225、2及-18℃。
[0083]
对实施例1得到的a类双酯型新戊基多元醇酯粗产品进行两级分子蒸馏精制,第一级分子蒸馏的操作条件如下:蒸发器温度为170℃,内冷器温度为10℃,绝对压力为2.4pa,刮膜速率为390min-1
。所得重组分(h1)的收率、酸值分别为92.05wt.%、0.08mgkoh/g。保持其它条件不变,在180℃的蒸发温度下对h1进行第二级分子蒸馏精制,得到a类双酯型新戊基多元醇酯精制产品,具有式(i-1)所示的结构:
[0084][0085]
所得a类双酯型新戊基多元醇酯精制产品在40℃及100℃时的运动粘度、粘度指数、倾点、酸值、色度、密度、开口闪点及收率分别为1423.03mm2/s、159.53mm2/s、230、-16℃、0.03mgkoh/g、3、1.05g/ml、251℃及90.44wt.%,可用作超高粘度指数(uhvi)的高性能润滑油基础油。
[0086]
实施例2
[0087]
b类双酯型新戊基多元醇酯粗产品的合成:将三羟甲基丙烷、己二酸及2-乙基己醇按1:3:3的摩尔比分两步加入反应体系,第一步加入摩尔比为3:3的2-乙基己醇与己二酸,先形成“半酯结构”;第二步再加入三羟甲基丙烷,形成“双酯结构”。第一、二步反应的反应温度均为170℃,反应时间分别为1h与4h,催化剂加量为反应物料总质量的1%,氮气流量为
2ml
·
min-1
·
g-1
,搅拌速度为800rpm。待反应结束后,在真空条件下通过抽滤实现超高粘度指数酯类基础油与锡锆复合氧化物催化剂的分离,得到b类双酯型新戊基多元醇酯的粗产品。
[0088]
在上述合成条件下制得的b类双酯型新戊基多元醇酯粗产品在40℃及100℃时的运动粘度、粘度指数、色度及倾点分别为201.12mm2/s、26.66mm2/s、168、2及-32℃。
[0089]
对实施例2得到的b类双酯型新戊基多元醇酯粗产品进行两级分子蒸馏精制,第一级分子蒸馏的操作条件如下:蒸发器温度为170℃,内冷器温度为10℃,绝对压力为2.4pa,刮膜速率为390min-1
。所得重组分(h1)的收率、酸值分别为92.17wt.%、0.08mgkoh/g。保持其它条件不变,在180℃的蒸发温度下对h1进行第二级分子蒸馏精制,得到b类双酯型新戊基多元醇酯的精制产品,具有式(i-2)所示的结构:
[0090][0091]
所得b类双酯型新戊基多元醇酯精制产品在40℃及100℃时的运动粘度、粘度指数、倾点、酸值、色度、密度、开口闪点及收率分别为217.31mm2/s、28.81mm2/s、172、-30℃、0.03mgkoh/g、3、1.04g/ml、250℃及90.17wt.%,可用作超高粘度指数(uhvi)的高性能润滑油基础油。
[0092]
实施例3
[0093]
c类双酯型新戊基多元醇酯粗产品的合成:将季戊四醇、己二酸、2-乙基己醇及正庚酸按1:1:1:3的摩尔比分两步加入反应体系,第一步加入摩尔比为1:1的2-乙基己醇与己二酸,先形成“半酯结构”;第二步再加入摩尔比为1:3的季戊四醇与正庚酸,形成“双酯结构”。第一步反应在160℃条件下反应1.5h;第二步反应先在170℃条件下反应1h,然后升温至180℃,反应6h。催化剂加量、氮气流量及搅拌速度分别为1.5wt.%、2ml
·
min-1
·
g-1
和800rpm。
[0094]
待反应结束后,在真空条件下通过抽滤实现高粘度酯类基础油与锡锆复合氧化物催化剂的分离,得到c类双酯型新戊基多元醇酯的粗产品。
[0095]
在上述合成条件下制得的c类双酯型新戊基多元醇酯粗产品在40℃及100℃时的运动粘度、粘度指数、色度及倾点分别为74.91mm2/s、13.27mm2/s、181、3及-49℃。
[0096]
对实施例3得到的c类双酯型新戊基多元醇酯粗产品进行两级分子蒸馏精制,第一级分子蒸馏的操作条件如下:蒸发器温度为170℃,内冷器温度为10℃,绝对压力为2.4pa,刮膜速率为390min-1
。所得重组分(h1)的收率、酸值分别为93.80wt.%、0.07mgkoh/g;保持其它条件不变,在180℃的蒸发温度下对h1进行第二级分子蒸馏精制,得到c类双酯型新戊基多元醇酯精制产品,具有式(i-3)所示的结构:
[0097][0098]
所得c类双酯型新戊基多元醇酯精制产品在40℃及100℃时的运动粘度、粘度指数、倾点、酸值、色度、密度、开口闪点及收率分别为89.12mm2/s、15.37mm2/s、184、-47℃、0.03mgkoh/g、3、1.03g/ml、270℃及91.67wt.%,可用作超高粘度指数(uhvi)的高性能润滑油基础油。
[0099]
c类双酯型新戊基多元醇酯(季戊四醇、己二酸、2-乙基己醇及正庚酸的摩尔比为10:13:13:27,运动粘度、粘度指数、倾点及色度则分别为106.29mm2/s(40℃)、16.74mm2/s(100℃)、172、-45℃和4)的表征:
[0100]
(1)
13
c-nmr分析:
[0101]
图1(a)与(b)分别为c类双酯型季戊四醇酯的
13
c-nmr谱及dept 135
°
谱,其中季碳原子在dept 135
°
谱中无信号峰,伯碳与叔碳原子在dept135
°
谱中呈现正峰,仲碳在dept 135
°
谱中呈现负峰。图2为利用chemdraw软件预测的c类双酯型季戊四醇酯的
13
c-nmr谱化学位移。
[0102]
根据文献
[1-3]
及图2对c类双酯型季戊四醇酯的碳核磁谱图进行识别,其中化学位移为0及77.3ppm的峰分别对应内标物si(ch3)4和溶剂cdcl3。a峰(174.0ppm)为己二酸羰基碳原子的特征峰,表明粗酯中的酸性物质以己二酸单2-乙基己酯为主。b(173.1、173.3、173.4ppm)、c(172.5、172.6ppm)处的峰分别为正庚酸及己二酸与季戊四醇反应生成的酯基碳原子的特征峰。d1(66.6ppm)与d2(62.0ppm)处的峰在dept 135
°
谱中为负值,且处于低场区,可判断其为与酯基氧原子相连的仲碳原子的信号峰,分别归属于2-乙基己醇酯及季戊四醇酯。伯碳峰的化学位移通常较小,因此可判断e峰(38.8ppm)为2-乙基己醇的叔碳原子峰。f峰(41.9ppm)在dept 135
°
谱中消失,且化学位移相对较小,可判断其为季戊四醇的季碳原子特征峰。g、h及i处的负峰处于高场区,可判断其为仲碳原子的特征峰,其中响应值较高的g1(34.1ppm)、g2(24.9ppm)、g3(28.8ppm)、g4(31.5ppm)及g5(22.5ppm)处的峰分别归属于正庚酸的1~5号碳原子;h1(30.4ppm)、h2(29.0ppm)、h3(23.7ppm)及h4(22.9ppm)处的峰分别归属于2-乙基己醇的1~4号碳原子;i1(33.6ppm)与i2(24.2ppm)处的峰分别归属于己二酸的1、2号碳原子。j峰(14.1ppm)与k峰(11.1ppm)位于高场区,可判断其为伯碳原子的特征峰,其中响应值较大的j峰归于正庚酸的甲基碳原子与2-乙基己醇的5号碳原子;k处的峰归属于2-乙基己醇的6号碳原子。与醚键相连的碳原子的化学位移约为70ppm,在图1中并未发现相似位移的信号峰,表明醚化反应在c类双酯型季戊四醇酯的合成过程中极少发生,原料中双季戊四醇的含量亦极少。
[0103]
(2)1h-nmr分析:
[0104]
图3为双酯型季戊四醇酯的1h-nmr谱。根据文献
[4,5]
及chemdraw软件预测的1h-nmr谱化学位移(图4),对氢核磁谱图进行识别,其中化学位移为0与7.30ppm的峰分别对应内标物si(ch3)4与溶剂cdcl3;a峰(0.88ppm)为甲基氢原子的特征峰;b峰(1.27ppm)与c峰
(1.64ppm)为亚甲基氢原子的特征峰;d峰(2.29ppm)为与酯基碳原子相连的亚甲基或次甲基的氢原子特征峰;e峰为与酯基氧原子相连的亚甲基的氢原子特征峰,e1峰(4.12ppm)、e2峰(3.97ppm)分别归属于季戊四醇酯及2-乙基己醇酯;b、c、d及e峰的化学位移逐渐增大,表明其逐渐靠近吸电子基团。g1峰(4.29ppm)与g2峰(4.26ppm)为未酯化羟基的氢原子特征峰,分别归属于2-乙基己醇及季戊四醇。与之相应,f1峰(3.52ppm)和f2峰(3.40ppm)为与羟基相连的亚甲基的氢原子特征峰,其峰面积占比极低,表明季戊四醇与2-乙基己醇的大部分羟基已被酯化。此外,与桥联型新戊基多元醇酯的f峰相比,c类双酯型季戊四醇酯的f峰较小,表明其羟基转化率更高。
[0105]
表2为c类双酯型季戊四醇酯的1h-nmr谱的定量分析结果,其中sm为非活泼氢原子占比的核磁测量结果,可根据mestrenova软件测量的峰面积计算得到;s
t
为非活泼氢原子占比的理论计算结果,可根据反应物的初始摩尔比(per:aa:eo:ha=10:13:13:27)计算得到,计算公式为s
t
=某构型的非活泼氢原子数/(总氢原子数-羧基氢原子数-羟基氢原子数)。结果显示非活泼氢的测量值与理论计算值基本吻合,表明谱图识别基本无误。其中,d峰面积占比(14.02%)约等于e1峰及e2峰的面积占比之和(10.85%+3.28%=14.13%),表明与酯基相连的亚甲基氢原子守恒;e1峰与e2峰的面积之比(10.85/3.28=3.23)略大于季戊四醇与2-乙基己醇的初始羟基摩尔比(40/13=3.08),表明季戊四醇的羟基更容易酯化,其羟基转化率相对更高。根据e1及e2峰的测量值与理论值之比可估算出c类双酯型季戊四醇酯合成反应的羟基转化率为x
oh
=(10.58%+3.28%)/(10.58%+3.44%)*100%=98.86%,与其羧基转化率(97.54%)较为接近。
[0106]
表2c类双酯型季戊四醇酯的1h-nmr谱的定量分析结果
[0107]
peakδ,ppmtypesm,mol.%s
t
,mol.%a0.88-ch320.9521.03b1.27-ch
2-34.9235.19c1.64-ch
2-15.8815.74d2.29o=c-ch-/o=c-ch
2-14.3914.02e1(per)4.12o=c-o-ch
2-10.5810.58e2(eo)3.97o=c-o-ch
2-3.283.44total4.12~0.88inactivehydrogen100100
[0108]
(3)gpc分析:
[0109]
表3为c类双酯型季戊四醇酯的分子量分布的分析。结果显示c类双酯型季戊四醇酯的数均分子量、重均分子量分别为1130、3972g/mol。
[0110]
表3 c类双酯型季戊四醇酯的gpc分析结果
[0111][0112][0113]
综上所述,由于更为合理的酯分子构型设计及合成方法的创新,本发明合成的双酯型新戊基多元醇酯的粘温性能极佳。由于粗产品中的水、有机酸、半酯与低沸点酯等非理想组分在分子蒸馏精制过程中被有效脱除,本发明合成的超高粘度指数酯类基础油的酸值
较低、稳定性较高。
[0114]
参考文献:
[0115]
[1]马楷,赵玉贞,梅莉.三种己二酸庚酸三羟甲基丙烷酯的性能对比[j].合成润滑材料,2016,43(2):7-10.
[0116]
[2]史伟,马楷,曹凯,等.一步法合成三羟甲基丙烷脂肪酸己二酸混合酯[j].当代化工,2016,45(1):31-33.
[0117]
[3]曹凯,罗玉兰.合成润滑油核磁共振谱解析[j].合成润滑材料,2013,40(3):9-11.
[0118]
[4]ji h.r,wang b.w,zhang x,et al.synthesis of levulinic acid-based polyol ester and its influence on tribological behavior as a potential lubricant[j].rsc advances,2015,5(122):100443-100451.
[0119]
[5]babij n.r,mccusker e.o,whiteker g.t,et al.nmr chemical shifts of trace impurities:industrially preferred solvents used in process and green chemistry[j].organic process research&development,2016,20(30):661-667.
[0120]
所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。