基于crispr/cas9体系编辑本氏烟基因的载体及构建方法和应用
技术领域
1.本发明涉及基因工程领域,特别是涉及一种基于crispr/cas9体系编辑本氏烟基因的载体和应用。
背景技术:
2.tylccnv是一种单组份双生病毒,在田间伴随着卫星dnaβ(tylccnb)。 tylccnv/tylccnb复合侵染烟草、番茄、辣椒等多种茄科作物,感染的作物产生明显的矮化、叶片卷曲、黄化等症状,严重导致作物减产,造成巨大的经济损失,危害农业生产。
3.我国大部分省市的番茄、烟草等种植区长期受到tylccnv/tylccnb复合侵染的危害。目前,在研究和生产中可用的抗双生病毒资源非常有限,而且病毒的快速进化与田间、温室作物上病毒病的复合侵染严重等都会导致抗双生病毒的种质资源不断丧失抗性。如番茄ty-1 介导的对番茄黄化曲叶病毒(tylcv)的抗性会被其他病毒所抑制,抗木薯花叶病害(由多种木薯花叶双生病毒引起)的木薯cmd2抗性基因在植物体细胞胚胎发生再生时会丢失。
4.此外,目前生产上缺乏有效的抗病毒药剂来防治双生病毒病害,因此在以预防病毒病害发生的基础上,选育抗病毒品种尤为重要。
技术实现要素:
5.本发明的目的是提供一种基于crispr/cas9体系编辑本氏烟基因的载体及构建方法和应用。通过设计针对tylccnv/tylccnb研究的模式植物本氏烟的感病基因nbbzip60的 grna,构建了利用编辑上述内源基因实现阻碍双生病毒侵染的农杆菌侵染性克隆载体。
6.为实现上述目的,本发明采用的技术方案具体如下:
7.一种基于crispr/cas9体系编辑本氏烟基因的载体的构建方法,其特征在于:所述本氏烟基因为nbbzip60基因,如seq id no:4所示;针对本氏烟nbbzip60基因编码区序列靠近 5’端的pam位点设计特异性靶向nbbzip60的grna,构建pcambia 1300-bkg-g1载体,简称bkg-g1载体。
8.具体而言,包括以下步骤:
9.(1)在本氏烟nbbzip60基因编码区序列靠近5’端搜寻pam位点并设计特异性靶向 nbbzip60的grna;
10.(2)根据所设计的靶点序列以及载体插入位置的酶切位点信息合成两条单链grna的 oligo片段,如seq id no:1-2所示,然后经退火后形成带有粘性末端酶切位点的互补双链;
11.(3)用限制性内切酶eco31i线性化bkg载体,纯化回收;
12.(4)用t4 dna连接酶连接线性化的bkg载体与双链化的grna;
13.(5)转化dh5α大肠杆菌,筛选鉴定阳性克隆,阳性克隆质粒测序,获得正确插入
grna 的bkg载体。
14.将上述构建方法获得的载体质粒转化eha105农杆菌,获得农杆菌侵染性克隆菌株。
15.本发明基于crispr/cas9体系编辑本氏烟基因的载体可以用于抵御依赖本氏烟寄主 nbbzip60完成复制侵染的植物病毒的侵染。
16.本发明基于crispr/cas9体系编辑本氏烟基因的载体可以通过在本氏烟的瞬时表达或遗传转化该载体获得稳定表达材料。
17.本发明基于crispr/cas9体系编辑本氏烟基因的载体可用于抵御双生病毒的侵染。
18.同现有技术相比,本发明的突出效果在于:
19.本发明基于crispr/cas9系统靶标并编辑本氏烟nbbzip60基因来抑制 tylccnv/tylccnb侵染。本氏烟nbbzip60基因是发明人研究团队鉴定出的与 tylccnv/tylccnb编码的致病因子βc1蛋白直接相关的基因,通过对上述基因的编辑阻碍病毒依靠其进行侵染从而达到抗病毒效果。
20.本发明构建出的抑制tylccnv/tylccnb侵染的载体bkg-g1具有以下特征和优势:
21.(1)对本氏烟的nbbzip60基因序列的进行定点编辑;
22.(2)与传统的育种方法相比能更快更有效的获得抗病材料;
23.(3)该载体编辑的nbbzip60基因是双生病毒tylccnv/tylccnb复制时所依赖的重要感病寄主基因,应用该载体可明显减轻tylccnv/tylccnb的侵染。
24.(4)该载体编辑nbbzip60基因后的本氏烟植物生长发育正常,没有明显的发育缺陷,有利于后代的繁育。
25.(5)该载体编辑nbbzip60基因的本氏烟植物可以作为研究双生病毒在寄主体内复制侵染的机理研究以及为抗病品种选育做参考。
26.(6)该载体可以应用到其他依赖nbbzip60基因的双生病毒的抗病研究中。
27.下面结合附图说明和具体实施例对本发明所述的基于crispr/cas9体系编辑本氏烟基因的载体及构建方法和应用作进一步说明。
附图说明
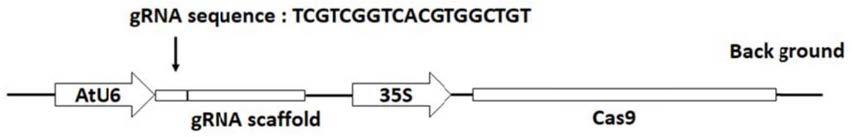
28.图1为基于crispr/cas9体系编辑本氏烟基因nbbzip60的载体构建示意图;
29.图2为针对nbbzip60基因编辑的载体对双生病毒tylccnv/tylccnb侵染的抑制效果。
30.(a)cas9-nbbzip60编辑的本氏烟植物的表型,与野生型wt相比较长势良好,没有明显的发育缺陷。
31.(b)编辑位点的测序结果,提取转入编辑载体的植物的dna,扩增编辑区域上下游200bp 后测序比对,发现pam位点附近存在nbbzip60序列的编辑,发生了碱基的插入或缺失,从而导致其后的翻译移码最终实现nbbzip60的敲除。
32.(c)tylccnv/tylccnb侵染野生型本氏烟和nbbzip60编辑的本氏烟7天之后的植物病毒症状。
33.(d)qpcr检测tylccnv/tylccnb的病毒积累水平。对图2c中的本氏烟系统叶片提取
dna,经qpcr分析与对照相比病毒dna水平的差异,25s rrna作为内参,**代表显著水平p<0.01。
具体实施方式
34.实施例1
35.结合图1所示,基于crispr/cas9体系编辑本氏烟基因的载体的构建方法,具体步骤如下:
36.(1)利用华中农业大学开发的crispr工具(http://crispr.hzau.edu.cn/crispr2/)在本氏烟nbbzip60基因编码区序列靠近5’端选定合适的pam位点并设计特异性的grna;因为插入到载体上的序列只有20个碱基,因此通过先合成两条引物,然后再退火成具有粘性末端的杂交双链。
37.grna-oligo a:tgattgtcgtcggtcacgtggctgt
38.grna-oligo b:aaacacagccacgtgaccgacgaca
39.(2)按照常规引物分别合成oligo a、b(如seq id no:1-2所示),然后将得到的引物经过退火得到双链dna,使用碧云天annealing buffer for dna oligos(d0251)的方法。将步骤(1)中所述的grna-oligo a/b退火合成双链,具体步骤如下:
40.①
将grna-oligo a/b干粉用ddh2o配置为50μm;
41.②
在pcr管中配置反应体系:
42.ddh2o4μlannealing buffer for dna oligo2μloligo a2μloligo b2μl总共10μl
43.③
用pcr仪器控温:95℃2分钟;每8秒下降0.1℃,降到25℃;4℃5分钟暂时存放;
44.④
退火产物用于后续的连接或者冻存于-20冰箱。
45.(3)bkg载体的酶切:
46.①
在pcr管中配置反应体系:
47.bkg载体质粒dna1000ng10
×
buffer g2μleco31i1μlddh2o补足到20μl总共20μl
48.②
将上述体系用移液器吸打混匀,于37℃孵育2小时。
49.③
将酶切产物跑胶,用胶回收试剂盒回收,后续的连接或者冻存于-20冰箱
50.(4)将获得的双链oligo dna和酶切的bkg载体通过t4连接酶连接。
51.10
×
t4dna ligase buffer1μlbkg载体(eco31i切)30ng双链oligo dna5μl
ddh2o补足到10μl
52.pcr仪控温22℃反应30分钟。
53.(5)将步骤(4)的连接产物转化dh5α大肠杆菌,菌液涂板于卡那霉素筛选的培养平板,37℃倒置培养过夜,挑选单菌落做菌落pcr鉴定阳性克隆;
54.鉴定引物:
55.f引物:m13f:gttgtaaaacgacggccag;(如seq id no:3所示)
56.r引物:grna-oligo b:aaacacagccacgtgaccgacgaca;
[0057]2×
taq mix10μlm13f(10μm)0.5μlgrna-oligo b(10μm)0.5μlddh2o9μl
[0058]
用10μl灭菌枪头蘸取一点单菌落到上述体系中,移液器吸打混匀
[0059]
pcr程序如下
[0060]
95℃5分钟;
[0061]
95℃30秒
[0062]
55℃30秒
[0063]
72℃30秒
[0064]
步骤2-4循环22次
[0065]
72℃5分钟
[0066]
4℃2分钟
[0067]
将pcr产物跑胶,筛选阳性克隆。
[0068]
(6)挑取阳性克隆单菌落置于含卡那霉素抗性的lb培养基中,37℃培养12-16个小时,提取质粒,并用m13f通用引物测序,至此得到加grna靶点的bkg-g1载体。
[0069]
实施例2
[0070]
(1)将实施例1所获得的载体质粒转化农杆菌:
[0071]
将测序正确的载体转化eha105农杆菌,取农杆菌eha105菌株50μl,质粒2μl,冻融法转化;卡那霉素+利福平抗平板筛选,挑选单菌落pcr鉴定阳性克隆,鉴定引物同实施例 1的步骤(5),将阳性农杆菌单菌落用卡那霉素+利福平lb液体培养基培养16-18小时;-80℃保存菌株。
[0072]
(2)将bkg-g1载体通过组培技术转化到本氏烟植物,对获得的阳性转化植株进行鉴定 (如图2a),提取dna,扩增nbbzip60编辑区域上下游200bp,然后测序鉴定,发现nbbzip60 序列pam位点附近存在碱基缺失或插入,表明nbbzip60确实能够被有效的编辑(如图2b),已编辑的植株后续收取种子,繁育后代。
[0073]
tylccnv/tylccnb侵染野生型本氏烟和nbbzip60编辑的本氏烟7天之后的植物病毒症状如图2c所示;qpcr检测tylccnv/tylccnb的病毒积累水平如图2d所示。
[0074]
本发明利用crispr-双生病毒编辑新技术,结合鉴定的关键植物新靶标,创制高抗双生病毒的遗传材料,具有高抗双生病毒的特性。该方法通过前沿的精细分子育种技术,通过 crispr敲除感病基因,获得的后代植物不含有转基因,在食品安全中更容易被大众接受,不仅在双生病毒病的绿色防控中具有重要的应用前景,也为其他抗性育种提供了案例。
[0075]
以上所述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明权利要求书确定的保护范围内。