1.本发明属于有机合成领域,具体涉及一种伊多塞班及其中间体的制备方法。
背景技术:
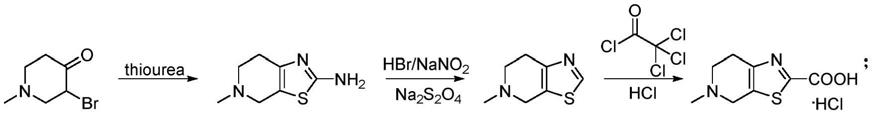
2.伊多塞班edoxaban(du-176)是口服的fxa抑制剂,临床开发用于中风预防。其中4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐是合成伊多塞班的重要中间体。目前合成该化合物的方法主要有以下三种:
[0003]
(1)以3-溴-1-甲基-哌啶-4-酮和硫脲为原料一步缩合合成4,5,6,7-四氢-5-甲基-2-氨基噻唑并[5,4-c]吡啶,其经桑德迈尔反应和脱卤反应生成4,5,6,7-四氢-5-甲基噻唑并[5,4-c]吡啶。该中间体再与三氯代乙酰氯经一系列转化生成最终的目标产物,
[0004][0005]
(2)以3-溴-1-甲基-哌啶-4-酮、硫粉和氨基腈为原料一步缩合合成4,5,6,7-四氢-5-甲基-2-氨基噻唑并[5,4-c]吡啶。其经桑德迈尔反应合成4,5,6,7-四氢-5-甲基-2-溴噻唑并[5,4-c]吡啶。该中间体再与甲酸苯酯经一系列转化生成最终的目标产物,
[0006][0007]
(3)首先通过方法(2)路径合成4,5,6,7-四氢-5-甲基-2-溴噻唑并[5,4-c]吡啶,其再与co2反应制备最终的目标产物,
[0008][0009]
然而,上述三种方法都存在步骤长,后处理麻烦,收率不高的问题(总收率基本小于40%),且三种方法都需要进行桑德迈尔反应,危险性较高,不易大规模生产。
技术实现要素:
[0010]
本发明的目的是克服现有技术的不足,提供一种改进的兼具步骤短、收率高、后处理简单和安全性好的制备4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐的方法。
[0011]
本发明同时还提供了一种伊多塞班的制备方法。
[0012]
为达到上述目的,本发明采用的一种技术方案是:
[0013]
一种4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐的制备方法,所述制备方法包括:
[0014]
(1)使3-溴-1-甲基-哌啶-4-酮和硫代草酰胺乙酯在碱存在下、在溶剂中、在反应温度为50-85℃下经缩合反应,生成5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙
酯;
[0015][0016]
(2)使5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯经水解,盐酸化,生成所述4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐;
[0017][0018]
根据本发明的一些优选方面,步骤(1)中,所述碱为选自碳酸钠、碳酸钾、氢氧化钠、氢氧化钾和三乙胺中的一种或多种的组合,在碱存在下进行该缩合反应能够显著提升反应收率。
[0019]
根据本发明的一些优选方面,步骤(1)中,控制所述缩合反应的反应时间为6-16h。
[0020]
根据本发明的一些优选且具体的方面,步骤(1)中,所述碱为选自碳酸钠、碳酸钾、氢氧化钠、氢氧化钾中的一种或多种的组合,所述缩合反应的反应时间为8-12h。
[0021]
根据本发明的一些优选且具体的方面,步骤(1)中,所述碱为三乙胺,所述缩合反应的反应时间为14-16h。
[0022]
根据本发明的一些优选方面,步骤(1)中,所述3-溴-1-甲基-哌啶-4-酮、所述硫代草酰胺乙酯和所述碱的投料摩尔比为1∶1-2∶0.1-0.5。进一步优选地,步骤(1)中,所述3-溴-1-甲基-哌啶-4-酮、所述硫代草酰胺乙酯和所述碱的投料摩尔比为1∶1-1.5∶0.1-0.3。
[0023]
根据本发明的一些优选方面,步骤(1)中,所述溶剂为醇类溶剂。
[0024]
根据本发明的一些优选方面,步骤(1)中,控制所述溶剂的添加量以使所述3-溴-1-甲基-哌啶-4-酮的浓度为0.5-2.0mmol/ml。根据本发明的一些优选且具体的方面,步骤(1)中,所述溶剂为醇类溶剂,控制所述醇类溶剂的添加量以使所述3-溴-1-甲基-哌啶-4-酮的浓度为0.5-2.0mmol/ml。
[0025]
根据本发明的一些优选且具体的方面,所述醇类溶剂为选自甲醇、乙醇和异丙醇中的一种或多种的组合。
[0026]
根据本发明的一些优选且具体的方面,步骤(1)中,使所述缩合反应在回流条件下进行。
[0027]
根据本发明的一些优选方面,步骤(2)中,使所述水解在氢氧化钠存在下进行。
[0028]
根据本发明的一些具体且优选的方面,步骤(2)中,通过添加氢氧化钠水溶液使水解反应进行。在本发明的一些实施方式中,所述氢氧化钠水溶液的浓度为2-10mol/l,进一步为4-10mol/l。根据本发明的一个具体方面,所述氢氧化钠水溶液的浓度为5mol/l。
[0029]
根据本发明的一些具体且优选的方面,步骤(2)中,先使5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯溶解在醇类溶剂中,然后添加氢氧化钠水溶液,水解反应。
[0030]
根据本发明的一些具体且优选的方面,步骤(2)中,使水解反应在回流条件下进行。
[0031]
根据本发明的一些具体且优选的方面,步骤(2)中,控制所述水解反应的反应时间为2-8h。
[0032]
根据本发明的一些优选方面,步骤(2)中,所述盐酸化通过添加盐酸并调节ph值至1-3。
[0033]
根据本发明的一些优选方面,步骤(2)中,控制所述盐酸化在低于5℃下进行。根据本发明的一个具体方面,步骤(2)中,控制所述盐酸化在冰浴条件下进行。
[0034]
根据本发明的一些优选方面,步骤(2)中,先使5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯溶解在醇类溶剂中,然后添加氢氧化钠水溶液,水解反应,反应完成后冷却至温室,蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质,水相在冰浴条件下用浓盐酸(质量分数36-38%)调解ph值至1-3,搅拌析出白色固体(可采用抽滤,干燥方式进行后处理即可,操作简单方便),即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐。
[0035]
根据本发明,3-溴-1-甲基-哌啶-4-酮和硫代草酰胺乙酯均可通过商购获得或者根据本领域常规方法制备而得。
[0036]
优选地,3-溴-1-甲基-哌啶-4-酮可按照如下方法制备:小于5℃条件下(例如冰浴条件),将1-甲基-哌啶-4-酮悬浮于乙醚80ml中,然后加入nbs(n-溴代琥珀酰亚胺)和醋酸铵搅拌反应后抽滤除去白色固体,滤液减压浓缩得到黄色油状液的即为3-溴-1-甲基-哌啶-4-酮。进一步地,在一个具体实施方式中,冰浴条件下,将1-甲基-哌啶-4-酮11.3g(100mmol)悬浮于乙醚80ml中,然后加入nbs 19.6g(110mmol)和醋酸铵0.77g(10mmol)搅拌反应4h后抽滤除去白色固体,滤液减压浓缩得到黄色油状液的即为3-溴-1-甲基-哌啶-4-酮(15.9g,纯度96.5%,产率83%)。
[0037]
本发明提供的又一技术方案:一种伊多塞班的合成方法,所述合成方法包括上述所述的4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐的制备方法。
[0038]
由于上述技术方案运用,本发明与现有技术相比具有下列优点:
[0039]
本发明基于现有制备4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐存在的缺陷,创新地提供了一种以以3-溴-1-甲基-哌啶-4-酮和硫代草酰胺乙酯为原料,经一步缩合合成中间体5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯,其再通过水解和盐酸化得到最终的目标产物。与现有文献方法(总收率基本小于40%)相比,该方法具有反应步骤短、收率高、后处理简单等优点,且反应过程可控,安全性较高,适于规模化生产。
附图说明
[0040]
图1为本发明实施例1制成的4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐的核磁谱图。
具体实施方式
[0041]
以下结合具体实施例对上述方案做进一步说明;应理解,这些实施例是用于说明本发明的基本原理、主要特征和优点,而本发明不受以下实施例的范围限制;实施例中采用的实施条件可以根据具体要求做进一步调整,未注明的实施条件通常为常规实验中的条件。
[0042]
下述实施例中未作特殊说明,所有原料均来自于商购或通过本领域的常规方法制
备而得。
[0043]
以下实例的反应过程如下反应式所示:
[0044][0045]
实施例1
[0046]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml乙醇中,加入碳酸钠(2mmol,212mg),在回流条件下反应10h。反应结束后冷却至室温,将不溶物滤除,滤液蒸干,加入碳酸氢钠水溶液洗至微碱性,然后用乙酸乙酯萃取、干燥、浓缩后,过柱分离得到5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯。
[0047]
其进一步溶解于10ml乙醇中,加入5mol/l naoh水溶液4ml(0.02mol),反应回流4h。冷却至室温后蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质后,水相在冰浴下用浓盐酸(37%)调节ph至2,搅拌析出白色固体,将白色固体抽滤出,干燥后所得白色固体即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐(产率67%,纯度98.5%,1.57g),其核磁谱图如图1所示。
[0048]
实施例2
[0049]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml乙醇中,加入naoh(2mmol,80mg),在回流条件下反应10h。反应结束后冷却至室温,将不溶物滤除,滤液蒸干,加入碳酸氢钠水溶液洗至微碱性,然后用乙酸乙酯萃取、干燥、浓缩后,过柱分离得到5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯。
[0050]
其进一步溶解于10ml乙醇中,加入5mol/l naoh水溶液4ml(0.02mol),反应回流4h。冷却至室温后蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质后,水相在冰浴下用浓盐酸(37%)调节ph至2,搅拌析出白色固体,将白色固体抽滤出,干燥后所得白色固体即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐(产率75%,纯度98.9%,1.76g)。
[0051]
实施例3
[0052]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml甲醇中,加入碳酸钠(2mmol,212mg),在回流条件下反应16h。反应结束后冷却至室温,将不溶物滤除,滤液蒸干,加入碳酸氢钠水溶液洗至微碱性,然后用乙酸乙酯萃取、干燥、浓缩后,过柱分离得到5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯。
[0053]
其进一步溶解于10ml乙醇中,加入5mol/l naoh水溶液4ml(0.02mol),反应回流4h。冷却至室温后蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质后,水相在冰浴下用浓盐酸(37%)调节ph至2,搅拌析出白色固体,将白色固体抽滤出,干燥后所得白色固体即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐(产率55%,纯度98.1%,1.29g)。
[0054]
实施例4
[0055]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml乙醇中,加入三乙胺(2mmol,0.28ml),在回流条件下反应16h。反应结束后冷却至室温,将不溶物滤除,滤液蒸干,加入饱和碳酸氢钠水溶液洗至微碱性,然后用乙酸乙酯萃取、
干燥、浓缩后,过柱分离得到5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯。
[0056]
其进一步溶解于10ml乙醇中,加入5mol/l naoh水溶液4ml(0.02mol),反应回流4h。冷却至室温后蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质后,水相在冰浴下用浓盐酸(37%)调节ph至2,搅拌析出白色固体,将白色固体抽滤出,干燥后所得白色固体即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐(产率65%,纯度98.5%,1.53g)。
[0057]
对比例1
[0058]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml乙醇中,在回流条件下反应10h。反应结束后冷却至室温,将不溶物滤除,滤液蒸干,加入碳酸氢钠水溶液洗至微碱性,然后用乙酸乙酯萃取、干燥、浓缩后,过柱分离得到5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯。
[0059]
其进一步溶解于10ml乙醇中,加入5mol/l naoh水溶液4ml(0.02mol),反应回流4h。冷却至室温后蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质后,水相在冰浴下用浓盐酸(37%)调节ph至2,搅拌析出白色固体,将白色固体抽滤出,干燥后所得白色固体即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐(产率28%,纯度95.8%,0.66g)。
[0060]
对比例2
[0061]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml乙醇中,在室温条件下反应10h。无明显5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯生成。
[0062]
对比例3
[0063]
将3-溴-1-甲基-哌啶-4-酮(10mmol,1.92g)和硫代草酰胺乙酯(11mmol,1.46g)溶于10ml乙醇中,加入碳酸钠(2mmol,212mg),在室温条件下反应10h。反应结束后,将不溶物滤除,滤液蒸干,加入碳酸氢钠水溶液洗至微碱性,然后用乙酸乙酯萃取、干燥、浓缩后,过柱分离得到5-甲基-4,5,6,7-四氢噻唑并[5,4-c]吡啶-2-羧酸乙酯。
[0064]
其进一步溶解于10ml乙醇中,加入5mol/l naoh水溶液4ml(0.02mol),反应回流4h。冷却至室温后蒸除溶剂后用水溶解残物,用乙酸乙酯/水萃取除去有机杂质后,水相在冰浴下用浓盐酸(37%)调节ph至2,搅拌析出白色固体,将白色固体抽滤出,干燥后所得白色固体即为4,5,6,7-四氢-5-甲基-噻唑并[5,4-c]吡啶-2-羧酸盐酸盐(产率15%,纯度97.6%,0.35g)。
[0065]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
[0066]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。