ether)其中的一种。其中,按重量份计,化合物a为100-110份,溶剂为2000-3000份;所述溶剂经过试剂纯化仪器干燥;
6.(2)引发剂的制备,分为两种方法:
7.(a)当步骤(1)中所用的溶剂为四氢呋喃(thf)溶剂时,先将混合溶液冷却至-78℃,然后在真空条件下向混合液中滴加正丁基锂(n-buli)60-160份,在-78℃下反应1-3小时,制备得到引发剂。
8.(b)当步骤(1)中所用的溶剂为其它溶剂时,在20-30℃真空条件下,直接向步骤(1)混合液中滴加正丁基锂(n-buli)60-160份,反应1-3小时,制备得到引发剂。
9.(3)真空条件下,向两口瓶中加入溶剂2000-3000份和六甲基环三硅氧烷(hexamethylcyclotrisiloxane,d3)单体600份;这里所述的溶剂同步骤(1);
10.如选用四氢呋喃(thf)为反应溶剂,则将体系温度冷却至-10到-15℃,然后取步骤(2-a)中制备的引发剂加入到体系中,保持-10到-15℃条件下搅拌反应1-2小时;
11.如反应溶剂为环己烷或其它溶剂,则用步骤(2-b)制备的引发剂加入到体系中,在室温下反应12-16小时。
12.(4)向步骤(3)反应体系中滴加预先蒸馏纯化过的二甲基一氯硅烷(chlorodimethylsilane)44-66份进行偶联终止反应,滴加结束后,室温条件下继续反应8-12小时。
13.(5)后处理:反应结束后,将反应液先进行减压蒸馏得到油状透明液体粗产物;然后在20000-30000份甲醇(meoh)中反复进行5-8次沉淀;再经过滤干燥,得到纯产物,产率大于70%。
14.(6)水解:将步骤(5)得到的产物在三氟乙酸(tfa)800-1200份或浓度为20%的氢氧化钠(naoh)水溶液1000-1600份/乙醇溶液400-800份中进行水解,时间8-13小时,得到羧基、胺基和二甲基硅端基官能化的聚二甲基硅氧烷。
15.其中,使用组分均按质量份计。
16.本发明中,所述溶剂优选:四氢呋喃(thf)和环己烷(cyclohexane)。
17.本发明中,所述用于制备功能化引发剂的化合物选自六甲基二硅氮烷、5-溴噻吩-2-甲酸甲酯、4-溴苯甲酸叔丁酯、4-溴-n,n-双(三甲基甲硅烷基)苯胺、5-溴-2-糠酸甲酯、4-溴-3-甲基苯甲酸叔丁酯及其衍生物中的一种。
18.本发明步骤(1)中,优选化合物a为100-110份。
19.本发明步骤(2)中,优选正丁基锂(n-buli)为60-160份(注:正丁基锂与化合物a的摩尔比要小于1)。
20.本发明中,用于合成引发剂的化合物为带有不同功能性结构的化合物,功能性结构的引入为聚合物功能化提供了条件。
21.本发明的有益效果:本发明方法为制备多种端基官能化聚二甲基硅氧烷提供了新的高效和便捷的合成途径。引发剂结构可以灵活设计,可实现多种端机功能化聚合物合成,室温下合成聚合物,大大降低了能耗,并且脱保护是在常压室温下进行,避免了过去通过高温和压力的方式,在工业化生产中降低能耗的同时提高了生产的安全性。
附图说明
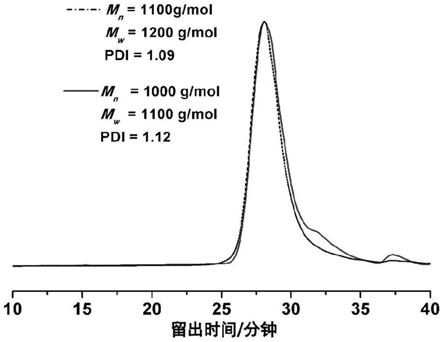
22.图1为实施例2中聚合物水解前后gpc的测试结果。其中,虚曲线为水解前,实曲线为水解后。
23.图2为实施例1中聚合物水解前后的氢核磁谱图。
24.图3为实施例2中聚合物水解前后的氢核磁谱图。
25.图4为实施例3中聚合物水解前后的氢核磁谱图。
具体实施方式
26.下面结合实施例对本发明作进一步描述。
27.实施例1
28.使用材料为:4-溴苯甲酸叔丁酯(tert-butyl-4-bromobenzoate)105份,环己烷(cyclohexane)溶剂5000份,正丁基锂(n-buli)140份,六甲基环三硅氧烷(hexamethylcyclotrisiloxane,d3)单体600份,二甲基一氯硅烷(chlorodimethylsilane)44份,三氟乙酸(tfa)溶液800份。
29.合成路线如下:
[0030][0031]
合成步骤为:
[0032]
(1)配制化合物溶液:在真空条件下,将化合物3-溴苯甲酸叔丁酯(tert-butyl-3-bromobenzoate)105份溶于试剂纯化仪器干燥过的环己烷(cyclohexane)溶剂2000份加入到两口反应瓶中,得到混合溶液。
[0033]
(2)引发剂的制备:在25℃真空条件下,直接向(1)混合液中滴加正丁基锂(n-buli)140份,反应2小时,制备得到引发剂。
[0034]
(3)真空条件下,向两口瓶中加入环己烷(cyclohexane)溶剂3000份和六甲基环三硅氧烷(hexamethylcyclotrisiloxane,d3)单体600份。取步骤(2)中制备好的引发剂加入到体系中,室温条件下搅拌反应16小时。
[0035]
(4)向(3)反应体系中滴加预先蒸馏纯化过的二甲基一氯硅烷(chlorodimethylsilane)44份进行偶联终止反应,滴加结束后,室温下继续反应10小时。
[0036]
(5)后处理:反应结束后,将反应液先进行减压蒸馏得到油状透明液体粗产物。然后在30000份甲醇(meoh)中反复进行5次沉淀。最后经过滤干燥,得到纯产物,产率大于70%。
buli)160份,四氢呋喃(thf)溶剂2650份,六甲基环三硅氧烷(hexamethylcyclotrisiloxane,d3)单体600份,二甲基一氯硅烷(chlorodimethylsilane)66份,三氟乙酸(tfa)溶液1200份。
[0051]
合成路线如下:
[0052][0053]
合成的步骤为:
[0054]
(1)配制化合物溶液:在真空条件下,将化合物六甲基二硅氮烷(hexamethyldisilazane)100份溶于试剂纯化仪器干燥过的四氢呋喃(thf)溶剂2500份加入到两口反应瓶中,得到混合溶液。
[0055]
(2)引发剂的制备:先将(1)混合溶液冷却至-78℃,然后在真空条件下向混合液中滴加正丁基锂(n-buli)60份,在-78℃下反应3小时,制备得到引发剂。
[0056]
(3)真空条件下,向两口瓶中加入四氢呋喃(thf)溶剂2000份和六甲基环三硅氧烷(hexamethylcyclotrisiloxane,d3)单体600份,然后冷却至-10℃。取步骤(2)中制备好的引发剂加入到体系中,保持体系在-10℃条件下搅拌反应2小时。
[0057]
(4)向(3)反应体系中滴加预先蒸馏纯化过的二甲基一氯硅烷(chlorodimethylsilane)66份进行偶联终止反应,滴加结束后,室温下继续反应12小时。
[0058]
(5)后处理:反应结束后,将反应液先进行减压蒸馏得到油状透明液体粗产物。然后在20000份甲醇(meoh)中反复进行8次沉淀。最后经过滤干燥,得到纯产物,产率大于70%。
[0059]
(6)水解:将以上(5步骤)得到的聚合物在三氟乙酸(tfa)溶液1200份中进行12小时水解,得到羧基和二甲基硅端基官能化的聚二甲基硅氧烷。
[0060]
用凝胶色谱仪gpc对实施例1-3中的聚合物进行水解前后分子量测试分析,表征结果如表1所示:
[0061]
表1
[0062][0063]
表1为实施例1-3聚合物水解前后gpc表征结果,图1为实施例2中聚合物gpc测试曲线,结合表1和图1的结果可以看到通过阴离子聚合得到的聚合物分布较窄,这符合阴离子聚合的典型特征。同时,聚合物水解前后分子量变化不大,说明在脱除端基保护基团过程中聚合物比较稳定。
[0064]
图2、图3、图4分别对应实施例1-3中聚合物水解前后的氢核磁谱图。从图2和图3中核磁结果看到,水解前聚合物端基保护基团的甲基信号峰(-ch3)的存在,对比水解后甲基信号峰消失了,说明水解后保护基团已完全脱除,该方法可以实现端基为羧基结构聚二甲基硅氧烷的合成。类似的,从图4中核磁结果看到,对比水解前后甲基(-ch3)信号峰化学位移处的积分显示水解后积分值减少了,说明水解后保护基团已完全脱除,进一步说明该方法可以实现端基为氨基结构聚二甲基硅氧烷的合成。要强调的是本发明只提供引发剂结构设计合成合成方法以及聚合物合成的过程,聚合物的性能研究在这里不进行赘述。
[0065]
以上所述为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明实质内容上所作的任何修改、等同替换和简单改进等,均应包含在本发明的保护范围之内。