六碳磷酸酯及其制备方法、维生素a酯化物的制备方法
技术领域
1.本发明涉及有机合成技术领域,具体涉及六碳磷酸酯及其制备方法、维生素a酯化物的制备方法。
背景技术:
2.维生素a是一种人体维持正常代谢和机能所必需的脂溶性维生素,且维生素a及其衍生物也是一类重要的药品。商品化的维生素a产品主要是维生素a酯化物,尤其以维生素a醋酸酯最为常见。
3.目前,basf公司开发的c
15
+c5路线是合成维生素a醋酸酯的主要方法之一。c
15
+c5合成路线具体如下:
4.该合成路线在可行性和安全性方面都具有优势,但要想获得较高的产品收率,需要采用碱度较高的碱,且反应温度需要控制在-35℃以下,反应条件苛刻,耗能较大,工业操作难度高。
5.cn 101219983 a公开了一种改进的维生素a乙酸酯的制备方法,针对basf公司的c
15
+c5合成路线进行了改进,是通过加入大量吡啶或类吡啶作为溶剂来提高反应收率,可以采用常见的碱金属醇盐在相对容易的操作条件下达到可工业化的水平,但大量使用吡啶或类吡啶溶剂又会增加生产成本和后处理的难度。
6.现有的维生素a醋酸酯c
15
+c5合成工艺大多是以c
15
磷酸酯作为witting-horner试剂,反应过程中c
15
磷酸酯需要过量添加,意味着不可避免要损失部分的c
15
磷酸酯,而c
15
磷酸酯的制备成本较高,相应会导致维生素a醋酸酯的生产成本高,而且c
15
磷酸酯存在着1,3位、1,4位和2,4位等双键异构体,在反应过程中需要消耗很大一部分强碱用于重排转位,会增加碱性废水的产生量。
7.因此,有必要开发一种更加经济、环保的维生素a酯化物制备方法。
技术实现要素:
8.本发明的目的之一在于提供一种六碳磷酸酯。
9.本发明的目的之二在于提供一种上述六碳磷酸酯的制备方法。
10.本发明的目的之三在于提供一种利用上述六碳磷酸酯制备维生素a酯化物的方法。
11.本发明所采取的技术方案是:
horner反应,即得六碳磷酸酯
21.优选的,步骤1)所述1-氯-2-甲基-4-羟基-2-丁烯、3-溴丙烯的摩尔比为1:1.1~1:1.5。
22.优选的,步骤1)所述溶剂为丙酮、四氢呋喃、甲苯中的至少一种。
23.优选的,步骤1)所述反应在-10℃~40℃下进行。
24.优选的,步骤2)所述化合物i、羧酸盐的摩尔比为1:1.0~1:1.5。
25.优选的,步骤2)所述羧酸盐为甲酸钠、乙酸钠中的至少一种。
26.优选的,步骤2)所述化合物i、相转移催化剂的摩尔比为1:0.03~1:0.05。
27.优选的,步骤2)所述相转移催化剂为苄基三乙基氯化铵、四甲基溴化铵、四甲基氯化铵、四乙基溴化铵、四乙基氯化铵、四丁基溴化铵、四丁基氯化铵中的至少一种。
28.优选的,步骤2)所述溶剂为丙酮、乙腈中的至少一种。
29.优选的,步骤2)所述酯化反应在溶剂回流的条件下进行,反应时间为5h~8h。
30.优选的,步骤2)所述酯化产物、碳酸钠的摩尔比为1:0.05~1:0.15。
31.优选的,步骤2)所述酯交换反应在60℃~65℃下进行,反应时间为3h~5h。
32.优选的,步骤2)所述化合物i、cucl、2,2,6,6-四甲基哌啶氧化物的摩尔比为1:0.01~0.30:0.01~0.20。
33.优选的,步骤2)所述氧化反应在20℃~50℃下进行,反应时间为2h~4h。
34.优选的,步骤3)所述化合物ii、亚甲基二磷酸四乙酯的摩尔比为1:1.1~1:1.3。
35.优选的,步骤3)所述亚甲基二磷酸四乙酯、碱催化剂的摩尔比为1:1.3~1:1.6。
36.优选的,步骤3)所述碱催化剂为甲醇钠、乙醇钠、叔丁醇钠中的至少一种。
37.优选的,步骤3)所述溶剂为四氢呋喃-甲苯混合溶剂。
38.优选的,步骤3)所述witting-horner反应在20℃~40℃下进行,反应时间为3h~5h。
39.一种维生素a酯化物的制备方法包括以下步骤:
40.1)进行和2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛的witting-horner反应,得到烯丙基保护的视黄醇;
41.2)脱除烯丙基保护的视黄醇中的烯丙基,再与酸酐或酰氯进行酰化反应,即得维生素a酯化物。
42.优选的,一种维生素a酯化物的制备方法包括以下步骤:
43.1)将2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛和碱催化剂分散在溶剂中,进行witting-horner反应,得到烯丙基保护的视黄醇;
44.2)将烯丙基保护的视黄醇分散在pdcl2的甲醇溶液中,进行脱除烯丙基反应,再加入酸催化剂、酸酐或酰氯,进行酰化反应,即得维生素a酯化物。
45.优选的,步骤1)所述2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛的摩尔比为1.0:1~1.3:1。
46.优选的,步骤1)所述碱催化剂的摩尔比为1:1.4~1:1.6。
47.优选的,步骤1)所述碱催化剂为甲醇钠、乙醇钠、叔丁醇钠中的至少一种。
48.优选的,步骤1)所述溶剂为四氢呋喃-甲苯混合溶剂。
49.优选的,步骤1)所述witting-horner反应在20℃~40℃下进行,反应时间为4h~5h。
50.优选的,步骤2)所述烯丙基保护的视黄醇、pdcl2的摩尔比为1:0.10~1:0.12。
51.优选的,步骤2)所述脱除烯丙基反应在20℃~60℃下进行,反应时间为4h~6h。
52.优选的,步骤2)所述酸催化剂为硫酸、磷酸、对甲苯磺酸中的至少一种。
53.本发明的有益效果是:本发明的六碳磷酸酯可以作为witting-horner试剂与2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛缩合后再与酸酐进行酰化反应制备维生素a酯化物,制备过程高效、经济、环保,适合进行大规模工业化应用。
54.具体来说:
55.1)本发明在制备六碳磷酸酯的过程中,每一步采用的工艺都达到了工业化的水平,工艺简洁连贯,操作简单,条件温和,收率良好,产品纯度高;
56.2)本发明获得的烯丙基为保护基的六碳磷酸酯容易活化,可以在较温和的条件下应用到维生素a醋酸酯的合成中,改变了惯常以c
15
磷酸酯作为witting-horner试剂的思维
定式,可以降低维生素a酯化物合成的经济成本,经济效益可观;
57.3)本发明摒弃了通过kornblum氧化或sommelet氧化来制备c5醛酯的传统工艺,以烯丙基为保护基的六碳磷酸酯可以在较温和条件下应用到维生素a酯化物的合成工艺中,有助于解决现有技术制备c5醛酯的过程中所出现的众多问题(尤其是环保方面),在温和条件下高效率完成工艺的同时,还可以降低高盐分、强碱性和难处理废水的处理量;
58.4)本发明的维生素a酯化物的制备过程具有高效、经济、环保的特点,具有重要的应用价值;
59.5)本发明在制备维生素a酯化物时将酰化步骤后置,可以增加产品的多样性和灵活性,不仅可以制备维生素a醋酸酯,而且还可以根据实际需要转变酰化试剂(例如:采用不同的酸酐作为酰化试剂),合成出更多种类的维生素a酯化物(例如:维生素a棕榈酸酯、维生素a丙酸酯等),甚至还可以以视黄醇为原料快捷地实现其各类衍生物的制取。
附图说明
60.图1为实施例1中的3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯的气相色谱图。
61.图2为实施例1中的3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯的gc-mc谱图。
62.图3为实施例1中的维生素a醋酸酯的液相色谱图。
具体实施方式
63.下面结合具体实施例对本发明作进一步的解释和说明。
64.实施例1中的1-氯-2-甲基-4-羟基-2-丁烯通过以下方法制备得到:将45g的1-氯-2-羟基-2-甲基-3-丁烯和1-氯-2-甲基-4-羟基-2-丁烯的混合物(该混合物的制备参考cn 112028740 a中的实施例1)、50g的水、25g带磺酸基团的强酸性离子交换树脂和50g的甲苯加入三口烧瓶,室温下搅拌4h,过滤,将滤液加入分液漏斗,静置分层,取上层有机相加入饱和na2co3溶液洗涤,减压蒸馏回收溶剂甲苯,得到41.8g的1-氯-2-甲基-4-羟基-2-丁烯(纯度:98%(气相检测),收率:91%)。
65.实施例1:
66.一种六碳磷酸酯,其制备方法包括以下步骤:
67.1)将41.8g的1-氯-2-甲基-4-羟基-2-丁烯分散在50g的无水丙酮中,再加入75g的无水碳酸钾,搅拌15min,再在0℃条件下缓慢滴加54g的3-溴丙烯,滴加完毕后升温至40℃反应,用薄层色谱检测直至反应完全,减压蒸馏回收溶剂丙酮,加入乙酸乙酯萃取,水洗数次,加入适量无水硫酸钠干燥有机相,减压蒸馏回收溶剂,得到53.8g的(命名:1-氯-2-甲基-4-烯丙氧基-2-丁烯;收率:95%);
68.2)将53.8g的1-氯-2-甲基-4-烯丙氧基-2-丁烯分散在50ml的乙腈中,再加入34g的乙酸钠和3g的苄基三乙基氯化铵,回流反应7h,冷却至室温,过滤,取滤液进行减压蒸馏回收溶剂,再加入150ml的乙酸乙酯萃取,水洗数次,减压蒸馏回收溶剂,得到64.0g的酯化物(纯度:93%(气相检测),收率:96%);
69.3)将64.0g的酯化物分散在200ml的甲醇中,再加入4.25g的碳酸钠,65℃反应4h,期间低沸副产物不断蒸出,减压蒸馏回收甲醇,过滤除去碳酸钠,再加入200ml的乙酸乙酯萃取,再水洗数次,减压蒸馏回收溶剂,得到47.5g的1-羟基-2-甲基-4-烯丙氧基-2-丁烯(纯度:91.6%(气相检测),收率:88%);
70.4)将47.5g的1-羟基-2-甲基-4-烯丙氧基-2-丁烯分散在150ml的丙酮中,再加入0.8g的cucl和1g的2,2,6,6-四甲基哌啶氧化物(tempo),再通入氧气室温反应4h,过滤回收cucl,减压蒸馏回收丙酮,再加入150ml的甲醇,并用150ml的石油醚分数次萃取出tempo,取石油醚层减压蒸馏回收溶剂,得到tempo,取甲醇层减压蒸馏回收溶剂,得到45.1g的(命名:4-烯丙氧基-2-甲基-2-丁烯醛,纯度:93%(气相检测),收率:89%);
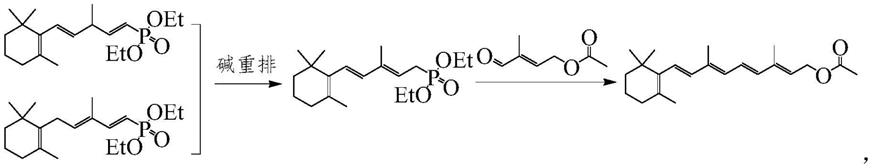
71.5)在氮气保护下,将26g的甲醇钠搅拌分散在60ml的四氢呋喃-甲苯混合溶剂(四氢呋喃、甲苯的体积比为2:1)中,降温至0℃,缓慢滴加106g的亚甲基二磷酸四乙酯,滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续搅拌1h,再滴加45.1g的4-烯丙氧基-2-甲基-2-丁烯醛(预先用15ml体积比2:1的四氢呋喃-甲苯混合溶剂分散),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续在20℃下反应3h,再向反应体系中加入质量分数1.0%的硫酸饱和nacl盐水溶液中和剩余的碱,再将反应液转入分液漏斗,静置分层,分出有机层,水层用50ml的甲苯萃取数次,合并有机相用50ml的饱和氯化钠水溶液洗涤数次,干燥,减压蒸馏回收溶剂,再进行蒸馏(约5mm hg柱),得到70.5g的六碳磷酸酯(命名:3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯,暗红色液体,纯度:98%(气相检测),收率:79%)。
72.3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯的合成反应如下:
[0073][0074]
3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯的气相色谱(岛津气相色谱仪gc-2014c)图如图1所示,gc-mc(气相色谱-质谱联用)谱图如图2所示。
[0075]
由图1可知:保留时间为8.924min的主峰指认为3-甲基-5-烯丙氧基-1,3-己二烯
磷酸酯,通过气相色谱面积归一法检测的纯度为98%。
[0076]
由图1和图2可知:本实施例确实合成得到了3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯。
[0077]
一种维生素a醋酸酯,其制备方法包括以下步骤:
[0078]
1)在氮气保护下,将20g的甲醇钠搅拌分散在60ml的四氢呋喃-甲苯混合溶剂(四氢呋喃、甲苯的体积比为2:1)中,降温至0℃,缓慢滴加70.5g的3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯(预先溶于30ml的甲苯),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续搅拌1h,再滴加44g的2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛(预先用15ml体积比2:1的四氢呋喃-甲苯混合溶剂分散),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续在20℃下反应3h,再向反应体系中加入质量分数1.0%的硫酸饱和nacl盐水溶液中和剩余的碱,再将反应液转入分液漏斗,静置分层,分出有机层,水层用50ml的甲苯萃取数次,合并有机相用50ml的饱和氯化钠水溶液洗涤数次,干燥,减压蒸馏回收溶剂,得到54.1g的烯丙基保护的视黄醇(淡黄油状液体,收率:74.5%);
[0079]
2)将54.1g的烯丙基保护的视黄醇分散在150ml的甲醇中,再加入3.2g的pdcl2,回流搅拌反应6h,过滤,取滤液减压蒸馏回收溶剂,得43.5g的暗黄色油状液体(收率:91.5%);
[0080]
3)将43.5g步骤2)的暗黄色油状液体和23.5g的乙酸酐混合,再搅拌加入1g的对甲苯磺酸,升温至65℃搅拌5h,降至室温,再加入100ml的水分层,取有机相,水层用乙酸乙酯萃取数次,合并有机相并减压蒸馏回收溶剂,向浓缩液中加入适量质量分数95%的乙醇,溶解后于50℃下搅拌10min,降温至-10℃保温结晶,过滤,得到29.9g的维生素a醋酸酯(纯度:99.1%(液相检测),收率:60.03%)。
[0081]
维生素a醋酸酯的液相色谱(岛津液相色谱仪lc-20at,色谱柱为inertsustain c
18
(250mm
×
4.6mm
×
5μm),流动相由甲醇和水按体积比3:2组成,检测波长为325nm)图如图3所示。
[0082]
由图3可知:根据标样比较法,证实该峰对应的化合物确实为维生素a醋酸酯。
[0083]
实施例2:
[0084]
一种六碳磷酸酯,其制备方法包括以下步骤:
[0085]
在氮气保护下,将39g的乙醇钠搅拌分散在80ml的四氢呋喃-甲苯混合溶剂(四氢呋喃、甲苯的体积比为2:1)中,降温至0℃,缓慢滴加128g的亚甲基二磷酸四乙酯,滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续搅拌1h,再滴加54.8g的4-烯丙氧基-2-甲基-2-丁烯醛(预先用15ml体积比2:1的四氢呋喃-甲苯混合溶剂分散;4-烯丙氧基-2-甲基-2-丁烯醛的制备过程和实施例1完全一样),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续在20℃下反应3h,再向反应体系中加入质量分数1.0%的硫酸饱和nacl盐水溶液中和剩余的碱,再将反应液转入分液漏斗,静置分层,分出有机层,水层用50ml的甲苯萃取数次,合并有机相用50ml的饱和氯化钠水溶液洗涤数次,干燥,减压蒸馏回收溶剂,再进行蒸馏(约5mm hg柱),得到84.1g的六碳磷酸酯
(暗红色液体,纯度:96%(气相检测),收率:75.9%)。
[0086]
一种维生素a醋酸酯,其制备方法包括以下步骤:
[0087]
1)在氮气保护下,将30g的乙醇钠搅拌分散在80ml的四氢呋喃-甲苯混合溶剂(四氢呋喃、甲苯的体积比为2:1)中,降温至0℃,缓慢滴加84.1g的3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯(预先溶于30ml的甲苯),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续搅拌1h,再滴加55g的2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛(预先用15ml体积比2:1的四氢呋喃-甲苯混合溶剂分散),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续在20℃下反应3h,再向反应体系中加入质量分数1.0%的硫酸饱和nacl盐水溶液中和剩余的碱,再将反应液转入分液漏斗,静置分层,分出有机层,水层用50ml的甲苯萃取数次,合并有机相用50ml的饱和氯化钠水溶液洗涤数次,干燥,减压蒸馏回收溶剂,得到64.9g的烯丙基保护的视黄醇(淡黄油状液体,收率:71.1%);
[0088]
2)将64.9g的烯丙基保护的视黄醇分散在150ml的甲醇中,再加入4.2g的pdcl2,回流搅拌反应5.5h,过滤,取滤液减压蒸馏回收溶剂,得50.3g的暗黄色油状液体(收率:88.3%);
[0089]
3)将50.3g步骤2)的暗黄色油状液体和27.5g的乙酸酐混合,再搅拌加入1.3g的对甲苯磺酸,升温至65℃搅拌5h,降至室温,再加入150ml的水分层,取有机相,水层用乙酸乙酯萃取数次,合并有机相并减压蒸馏回收溶剂,向浓缩液中加入适量质量分数95%的乙醇,溶解后于50℃下搅拌10min,降温至-10℃保温结晶,过滤,得到33.0g的维生素a醋酸酯(纯度:99.0%(液相检测),收率:57.2%)。
[0090]
实施例3:
[0091]
一种六碳磷酸酯,其制备方法包括以下步骤:
[0092]
在氮气保护下,将43g的叔丁醇钠搅拌分散在80ml的四氢呋喃-甲苯混合溶剂(四氢呋喃、甲苯的体积比为2:1)中,降温至0℃,缓慢滴加102.8g的亚甲基二磷酸四乙酯,滴加过程中控制反应体系温度不超过20℃,40min滴加完毕,继续搅拌1.5h,再滴加41g的4-烯丙氧基-2-甲基-2-丁烯醛(预先用15ml体积比2:1的四氢呋喃-甲苯混合溶剂分散;4-烯丙氧基-2-甲基-2-丁烯醛的制备过程和实施例1完全一样),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续在20℃下反应3h,再向反应体系中加入质量分数1.0%的硫酸饱和nacl盐水溶液中和剩余的碱,再将反应液转入分液漏斗,静置分层,分出有机层,水层用50ml的甲苯萃取数次,合并有机相用50ml的饱和氯化钠水溶液洗涤数次,干燥,减压蒸馏回收溶剂,再进行蒸馏(约5mm hg柱),得到64.8g的六碳磷酸酯
(暗红色液体,纯度:96.8%(气相检测),收率:78.6%)。
[0093]
一种维生素a醋酸酯,其制备方法包括以下步骤:
[0094]
1)在氮气保护下,将31.7g的叔丁醇钠搅拌分散在50ml的四氢呋喃-甲苯混合溶剂(四氢呋喃、甲苯的体积比为2:1)中,降温至0℃,缓慢滴加64.8g的3-甲基-5-烯丙氧基-1,3-己二烯磷酸酯(预先溶于25ml的甲苯),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续搅拌1h,再滴加40.4g的2-甲基-4-(2,6,6-三甲基-1-环己烯-1-基)-2-丁烯-1-醛(预先用15ml体积比2:1的四氢呋喃-甲苯混合溶剂分散),滴加过程中控制反应体系温度不超过20℃,30min滴加完毕,继续在20℃下反应3h,再向反应体系中加入质量分数1.0%的硫酸饱和nacl盐水溶液中和剩余的碱,再将反应液转入分液漏斗,静置分层,分出有机层,水层用50ml的甲苯萃取数次,合并有机相用50ml的饱和氯化钠水溶液洗涤数次,干燥,减压蒸馏回收溶剂,得到50.1g的烯丙基保护的视黄醇(淡黄油状液体,收率:75.3%);
[0095]
2)将50.1g的烯丙基保护的视黄醇分散在200ml的甲醇中,再加入3.2g的pdcl2,回流搅拌反应5.5h,过滤,取滤液减压蒸馏回收溶剂,得39.3g的暗黄色油状液体(收率:89.4%);
[0096]
3)将39.3g步骤2)的暗黄色油状液体和21.6g的乙酸酐混合,再搅拌加入1.5g的对甲苯磺酸,升温至65℃搅拌5h,降至室温,再加入150ml的水分层,取有机相,水层用乙酸乙酯萃取数次,合并有机相并减压蒸馏回收溶剂,向浓缩液中加入适量质量分数95%的乙醇,溶解后于50℃下搅拌10min,降温至-10℃保温结晶,过滤,得到26.9g的维生素a醋酸酯(纯度:99.3%(液相检测),收率:59.7%)。
[0097]
对比例:
[0098]
分别用酰氧基(参考cn 1097414 a)、吡喃基或硅醚基为保护基替代烯丙基保护基进行反应,对比不同保护基对合成六碳磷酸酯和维生素a醋酸酯的影响,测试结果如下表所示:
[0099]
表1不同保护基对合成六碳磷酸酯和维生素a醋酸酯的影响
[0100]
[0101][0102]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。