1.本发明涉及相变材料技术领域,具体涉及一种相变材料组合物、相变材料及其制备方法和应用。
背景技术:
2.无机水合盐相变储能材料作为一种新型节能储能材料,利用其潜热储能、释能,可以解决能量时空不匹配的问题,从而做到能量的循环再生利用,提高能源利用率。
3.无机水合盐相变储能材料在高温时吸热,将外界多余热量以潜热的形式存储,在环境温度降低时,将存储的热能再次释放至环境中,达到适时、自动调控环境温度的目的,维持环境温度相对稳定。其优异的温度调节和储能特性使其在很多领域如太阳能利用、建筑隔热保温、电子器件的热保护、食品保鲜等得到了广泛的应用。
4.然而,单纯的无机水合盐在相变过程中存在相分离、过冷度大、泄漏不稳定等问题。因此,如何减少相分离、降低腐蚀性以及提高材料的热性能对于相变材料的实际应用至关重要。
5.为了解决上述问题研究者提出了很多方案,比如通过引入增稠剂来增大体系的粘度,降低相分离程度,同时抓取水分,不让水分子逃离无机盐。然而,现有常用的增稠剂多为有机高分子,在高温等因素下,受储藏环境等影响,易于分解,分解后导致粘度降低,从而导致材料性能下降。
6.cn112680197a公开了一种无机水合盐相变材料及其制备方法,该方法通过引入水、多元醇等多羟基小分子破坏pva、pssa的分子内氢键,降低其结晶度,实现连续的低温热塑加工。
7.然而,前述方法制备得到的相变材料单位体积的焓值较低,而且仍然具有较大的过冷度。
8.因此,开发出一种能够有效抑制相分离、降低过冷度且单位质量热焓值高的相变材料,具有重要意义。
技术实现要素:
9.本发明的目的是为了克服现有技术相变材料存在单位质量热焓值较低、相分离严重、过冷度大的缺陷。
10.为了实现上述目的,本发明第一方面提供一种相变材料组合物,该组合物中含有两者以上混合保存或各自独立保存的以下组分:
11.90
‑
98重量%的无机水合盐、1
‑
8重量%的缓释型成核剂、0.2
‑
5重量%的增稠剂和0.2
‑
3重量%的缓蚀剂;
12.所述缓释型成核剂中含有活性成分以及包裹在所述活性成分表面的亲水型气凝胶,所述活性成分选自十二水合磷酸氢二钠、焦磷酸钠十水合物、硅酸钠、十水合硼酸钠、十水合碳酸钠、八水合氢氧化钡、六水合氯化锶、硼砂、六水氯化镁中的至少一种;在所述缓释
型成核剂中,所述亲水型气凝胶与所述活性成分的含量质量比为1:8
‑
30。
13.本发明第二方面提供一种制备相变材料的方法,该方法包括:将第一方面所述的组合物中的各组分进行混合。
14.本发明第三方面提供一种由第二方面所述的方法制备得到的相变材料。
15.本发明第四方面提供第三方面所述的相变材料在电器中的应用。
16.本发明第五方面提供第三方面所述的相变材料在厨房电器中的应用。
17.本发明采用亲水型气凝胶包覆水合盐活性成分形成缓释型成核剂,将该缓释型成核剂应用于相变材料中,能够有效抑制相变材料的相分离,获得单位质量热焓值高、过冷度低的相变材料。
附图说明
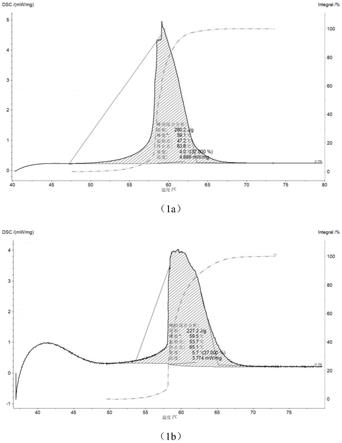
18.图1是实施例1制备得到的相变材料的初始单位质量热焓值dsc曲线图和循环12000次后的单位质量热焓值dsc曲线图;
19.图2是实施例2制备的相变材料的过冷度时间
‑
温度曲线。
具体实施方式
20.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
21.需要说明的是,在本发明的各方面中,针对各方面中的相同的组分,本发明仅在其中一方面中描述一次而不重复进行描述,本领域技术人员不应理解为对本发明的限制。
22.需要说明的是,在本发明中,未说明的原料均可由一般途径的商购获得,本领域技术人员可以根据需要进行采购,在本发明中,不再一一赘述。
23.本发明中,未作相反说明的情况下,所述常温或室温均表示25
±
2℃。
24.如前所述,本发明的第一方面提供了一种相变材料组合物,该组合物中含有两者以上混合保存或各自独立保存的以下组分:
25.90
‑
98重量%的无机水合盐、1
‑
8重量%的缓释型成核剂、0.2
‑
5重量%的增稠剂和0.2
‑
3重量%的缓蚀剂;
26.所述缓释型成核剂中含有活性成分以及包裹在所述活性成分表面的亲水型气凝胶,所述活性成分选自十二水合磷酸氢二钠、焦磷酸钠十水合物、硅酸钠、十水合硼酸钠、十水合碳酸钠、八水合氢氧化钡、六水合氯化锶、硼砂、六水氯化镁中的至少一种;在所述缓释型成核剂中,所述亲水型气凝胶与所述活性成分的含量质量比为1:8
‑
30。
27.优选地,以所述组合物的总重量为基准,所述组合物中含有90
‑
96重量%无机水合盐、2
‑
6重量%缓释型成核剂、1
‑
3重量%增稠剂和0.5
‑
2重量%缓蚀剂。
28.优选地,在所述缓释型成核剂中,所述亲水型气凝胶与所述活性成分的含量质量比为1:20
‑
30。发明人发现,采用该优选情况下的具体实施方式,能够获得循环性能更优异、过冷度更低的相变材料。
29.优选地,所述亲水型气凝胶的比表面积为100
‑
600m2/g,孔隙率为80
‑
99%,平均孔
径为1
‑
100nm,密度为1
‑
100kg/m3。
30.更优选地,所述亲水型气凝胶的比表面积为200
‑
600m2/g,孔隙率为85
‑
95%,平均孔径为50
‑
80nm,密度为5
‑
40kg/m3。
31.优选地,所述亲水型气凝胶选自石墨烯气凝胶、纳米二氧化硅气凝胶、氮化硅气凝胶、氮化硼气凝胶和mxene气凝胶中的至少一种。
32.优选地,所述无机水合盐选自三水合醋酸钠、六水合氯化钙、十水合氢氧化钡、七水合硫酸镁中的至少一种。
33.优选地,所述缓蚀剂选自苯骈三氮唑、巯基苯骈噻唑、九水偏硅酸钠、甲基苯骈三氮唑、聚天冬氨酸中的至少一种。
34.优选地,所述增稠剂选自明胶、黄原胶、羧甲基纤维素钠、甲基纤维素、聚丙烯酰胺、聚丙烯酸钠、聚乙二醇、聚乙烯醇中的至少一种。
35.优选地,所述缓释型成核剂由包括以下步骤的方法制备得到:
36.(a)将活性成分进行加热处理ii,得到熔融活性成分;
37.(b)将亲水型气凝胶与所述熔融活性成分进行接触混合,得到平均粒径为0.002
‑
15mm的缓释型成核剂。
38.优选地,在步骤(a)中,所述加热处理ii的条件至少包括:温度为40
‑
80℃,时间为0.5
‑
2h。
39.根据一种特别优选的具体实施方式,在步骤(b)中,所述接触混合为真空循环浸渍。发明人发现,采用该优选情况下的具体实施方式,能够显著缩短浸渍时间,提高浸渍效率,从而获得性能更优异的相变材料。
40.更优选地,在步骤(b)中,所述接触混合的条件至少包括:真空度为10
‑
100pa,温度为40
‑
80℃,时间为4
‑
6h。
41.优选地,在步骤(b)中,本发明中所述真空循环浸渍的操作方法包括以下步骤:
42.(i)第一容器、第二容器和真空泵依次连接,形成循环体系;
43.(ii)将活性成分通入所述第一容器中,亲水型气凝胶通入所述第二容器中并密封,形成真空度为10
‑
100pa的负压条件;
44.(iii)在温度为40
‑
80℃下,启动真空泵,将所述活性成分抽入第二容器中,流经所述第二容器后进入真空泵并返回所述第一容器中,循环4
‑
6h后,倒出多余的活性成分。
45.优选地,在步骤(b)中,该方法还包括:将所述接触混合后得到的产物进行粉碎,以得到平均粒径为0.002
‑
15mm的缓释型成核剂。
46.本发明中可以采用本领域已知的方法对所述缓释型成核剂进行粉碎,以得到平均粒径为0.002
‑
15mm的缓释型成核剂,示例性地,所述粉碎处理可以采用球磨机、高速粉碎机、便携式粉碎机中的至少一种。
47.如前所述,本发明的第二方面提供了一种制备相变材料的方法,该方法包括:将第一方面所述的组合物中的各组分进行混合。
48.优选地,所述将组合物中的各组分进行混合的操作包括以下步骤:
49.(1)将无机水合盐进行加热处理i,得到熔融无机水合盐;
50.(2)将缓蚀剂与所述熔融无机水合盐进行第一接触混合,得到第一混合物料;
51.(3)将所述第一混合物料、增稠剂和缓释型成核剂进行第二接触混合。
52.优选地,在步骤(1)中,所述加热处理i的条件至少包括:温度为40
‑
90℃,时间为0.5
‑
6h。
53.优选地,在步骤(2)中,所述第一接触混合的条件至少包括:搅拌速度为50
‑
500rpm,温度为40
‑
90℃,时间为0.5
‑
6h。
54.优选地,在步骤(3)中,所述第二接触混合的条件至少包括:搅拌速度为50
‑
500rpm,温度为40
‑
90℃,时间为0.5
‑
6h。
55.如前所述,本发明的第三方面提供了一种由第二方面所述的方法制备得到的相变材料。
56.如前所述,本发明的第四方面提供了第三方面所述的相变材料在电器中的应用。
57.如前所述,本发明的第五方面提供了第三方面所述的相变材料在厨房电器中的应用。
58.以下将通过实施例对本发明进行详细描述。以下实例中,在没有特别说明的情况下,使用的各种原料均为市售品。
59.亲水型气凝胶:石墨烯气凝胶
‑
1,比表面积为480m2/g,孔隙率为92%,平均孔径为10
‑
20nm,密度为17kg/m3,购自南京先丰纳米公司;
60.亲水型气凝胶:气相纳米二氧化硅气凝胶粉末,比表面积为480m2/g,孔隙率为90%,密度为30kg/m3,购自湖北汇富纳米材料公司;
61.无机水合盐:三水合醋酸钠;
62.缓蚀剂:九水偏硅酸钠;
63.缓蚀剂:苯骈三氮唑;
64.增稠剂:羧甲基纤维素钠;
65.增稠剂:甲基纤维素;
66.十二水合磷酸氢二钠、焦磷酸钠十水合物、三水合醋酸钠、九水偏硅酸钠、羧甲基纤维素钠、苯骈三氮唑、甲基纤维素均为分析纯试剂。
67.制备例1:制备缓释型成核剂s1
68.(a)在60℃下,将1000g的十二水合磷酸氢二钠加热熔融1h,得到熔融十二水合磷酸氢二钠1000g;
69.(b)在60℃下,将10g的石墨烯气凝胶
‑
1置于真空袋中,在真空度为20pa下,将前述得到的全部熔融十二水合磷酸氢二钠用真空泵抽入真空袋中,经过6h后,倒出多余的熔融十二水合磷酸氢二钠750g(石墨烯气凝胶
‑
1与熔融十二水合磷酸氢二钠的用量质量比为1:25),冷却后得到固体物料,在40℃下,将所述固体物料采用高速粉碎机进行多次粉碎,每次粉碎时间不超过40s,得到平均粒径为5mm的固体物料,并将粉碎后的所述固体物料在球磨机中球磨30min,得到平均粒径为0.1mm的缓释型成核剂s1。
70.制备例2:制备缓释型成核剂s2
71.(a)在90℃下,将1000g的焦磷酸钠十水合物加热熔融0.5h,得到熔融焦磷酸钠1000g;
72.(b)在90℃下,将40g的石墨烯气凝胶
‑
1置于真空袋中,在真空度为80pa下,将前述得到的全部熔融焦磷酸钠用真空泵抽入真空袋中,经过6h后,倒出多余的熔融焦磷酸钠十水合物200g(石墨烯气凝胶
‑
1与熔融焦磷酸钠的用量质量比为1:20),冷却后得到固体物
料,将所述固体物料采用高速粉碎机进行多次粉碎,得到平均粒径为5mm的缓释型成核剂s2。
73.制备例3:制备缓释型成核剂s3
74.(a)在50℃下,将2000g的十二水合磷酸氢二钠加热熔融2h,得到熔融十二水合磷酸氢二钠2000g;
75.(b)在80℃下,将60g的石墨烯气凝胶
‑
1置于真空袋中,在真空度为100pa下,将前述得到的全部熔融十二水合磷酸氢二钠用真空泵抽入真空袋中,经过6h后,倒出多余的熔融十二水合磷酸氢二钠800g(石墨烯气凝胶
‑
1与十二水合磷酸氢二钠的用量质量比为1:20),冷却后得到固体物料,在40℃下,将所述固体物料采用高速粉碎机进行多次粉碎,得到平均粒径为8mm的缓释型成核剂s3。
76.制备例4:制备缓释型成核剂s4
77.本制备例按照与制备例1相似的方法制备缓释型成核剂,所不同的是,在步骤(b)中,用等质量的气相纳米二氧化硅气凝胶粉末替换石墨烯气凝胶
‑
1。
78.其余步骤均与制备例1相同。
79.得到平均粒径为0.1m的缓释型成核剂s4。
80.制备例5:制备缓释型成核剂s5
81.本制备例按照与制备例1相似的方法制备缓释型成核剂,所不同的是,在步骤(b)中,应用的石墨烯气凝胶
‑
1为25g,其中,石墨烯气凝胶
‑
1与熔融十二水合磷酸氢二钠的用量质量比为1:10。
82.其余步骤均与制备例1相同。
83.得到平均粒径0.1mm的缓释型成核剂s5。
84.对比制备例1:制备缓释型成核剂ds1
85.本对比制备例按照与制备例1相似的方法制备缓释型成核剂,所不同的是,在步骤(b)中,应用的石墨烯气凝胶
‑
1为50g,其中,石墨烯气凝胶
‑
1与十二水合磷酸氢二钠的用量质量比1:5。
86.其余步骤均与制备例1相同。
87.得到平均粒径为0.1mm的缓释型成核剂ds1。
88.对比制备例2:制备缓释型成核剂ds2
89.本对比制备例按照与制备例1相似的方法制备缓释型成核剂,所不同的是,在步骤(b)中,用等质量的分子筛替换石墨烯气凝胶
‑
1。
90.其余步骤均与制备例1相同。
91.得到平均粒径为20mm的缓释型成核剂ds2。
92.实施例1
93.本实施例提供一种制备相变材料的方法,该方法包括以下步骤:
94.(1)在75℃下,将98g的三水合醋酸钠加热熔融1h,得到熔融三水合醋酸钠;
95.(2)在75℃下,将1g的九水偏硅酸钠与前述得到的全部熔融三水合醋酸钠在500rpm下搅拌混合2h得到第一混合物料;
96.(3)在75℃下,将2g的羧甲基纤维素钠、5g的缓释型成核剂s1和前述得到的第一混合物料在500rpm下搅拌混合4h,得到相变材料a1。
97.实施例2
98.本实施例提供一种制备相变材料的方法,该方法包括以下步骤:
99.(1)在60℃下,将98g的三水合醋酸钠加热熔融2h,得到熔融三水合醋酸钠;
100.(2)在70℃下,将1g的苯骈三氮唑与前述得到的全部熔融三水合醋酸钠在400rpm下搅拌混合2h得到第一混合物料;
101.(3)在60℃下,将1.5g的甲基纤维素、2.5g的缓释型成核剂s2和前述得到的第一混合物料在450rpm下搅拌混合4h,得到相变材料a2。
102.实施例3
103.本实施例提供一种制备相变材料的方法,该方法包括以下步骤:
104.(1)在80℃下,将98g的三水合醋酸钠加热熔融1h,得到熔融三水合醋酸钠;
105.(2)在60℃下,将1.5g的九水偏硅酸钠与前述得到的全部熔融三水合醋酸钠在350rpm下搅拌混合3h得到第一混合物料;
106.(3)在70℃下,将3g的羧甲基纤维素钠、5.5g的缓释型成核剂s3和前述得到的第一混合物料在400rpm下搅拌混合5h,得到相变材料a3。
107.实施例4
108.本实施例按照与实施例1相似的方法制备相变材料,所不同的是,在步骤(3)中,用等质量的缓释型成核剂s4替换缓释型成核剂s1。
109.其余步骤均与实施例1相同,得到相变材料a4。
110.实施例5
111.本实施例按照与实施例1相似的方法制备相变材料,所不同的是,在步骤(3)中,用等质量的缓释型成核剂s5替换缓释型成核剂s1。
112.其余步骤均与实施例1相同,得到相变材料a5。
113.对比例1
114.本对比例按照与实施例1相似的方法制备相变材料,所不同的是,在步骤(3)中,用等质量的缓释型成核剂ds1替换缓释型成核剂s1。
115.其余步骤均与实施例1相同,得到相变材料da1。
116.对比例2
117.本对比例按照与实施例1相似的方法制备相变材料,所不同的是,在步骤(3)中,用等质量的缓释型成核剂ds2替换缓释型成核剂s1。
118.其余步骤均与实施例1相同,得到相变材料da2。
119.对比例3
120.本对比例按照与实施例1相似的方法制备相变材料,所不同的是,在步骤(3)中,用等质量的十二水合磷酸氢二钠替换缓释型成核剂s1。
121.其余步骤均与实施例1相同,得到相变材料da3。
122.测试例
123.分别检测实施例和对比例制备得到相变材料的单位质量热焓值、过冷度,以及循环12000次后的单位质量热焓值,并计算单位质量热焓值衰减率,具体结果见表1。
124.其中,单位质量热焓值采用dsc差示扫描量热仪进行测试;
125.循环12000次后的单位质量热焓值的检测方法为:将相变材料装入循环测试装置
中,使其经过完全熔融
‑
结晶12000次后,通过dsc差示扫描量热仪测试其单位质量热焓值;
126.单位质量热焓值衰减率的计算公式为:
127.其中,δh
n
为循环n次后的单位质量热焓值,kj/kg;
128.δh0为初始单位质量热焓值,kj/kg;
129.过冷度的测试方法为:将100g相变材料放置于65℃的烘箱中后,在相变材料的中间位置放置热电偶,自然冷却到40℃,获得相变材料的时间
‑
温度曲线,曲线的拐点与温度上升后的最高点之间的差值为过冷度。
130.表1
[0131][0132]
从表1可以看出,采用本发明提供的相变材料组合物能够有效抑制相变材料的相分离,获得单位质量热焓值高、过冷度低的相变材料。
[0133]
本发明示例性地提供了实施例1制备得到的相变材料的初始单位质量热焓值dsc曲线图和循环12000次后的单位质量热焓值dsc曲线图,实施例2制备的相变材料的过冷度测试曲线,分别见图1和图2。
[0134]
其中,图1是实施例1制备得到的相变材料的初始单位质量热焓值dsc曲线图和循环后的单位质量热焓值dsc曲线图,图(1a)是实施例1制备得到的相变材料的初始单位质量热焓值dsc曲线图,图(1b)是实施例1制备得到的相变材料循环12000次后的单位质量热焓值dsc曲线图;图2是实施例2制备的相变材料的过冷度时间
‑
温度曲线。
[0135]
从图1和图2中可以看出,本发明实施例1制备得到的相变材料循环12000次后,仍然具有较高的单位质量热焓值,本发明实施例2制备得到的相变材料过冷度为3℃。
[0136]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。