通过离子催化剂低聚和加氢异构化由nao产生的基础油
背景技术:
1.为了增加汽车的燃料经济性,汽车制造商正在开发使用非常低黏度的发动机油(ow
‑
xx发动机油,包括ow
‑
8和ow
‑
12,其超过市场上已有的黏度等级)来运行的更高效率内燃机。预计未来对这种低黏度发动机油的需求将迅速增长。
2.为了制造这种用于高效率发动机的专业低黏度润滑剂,除了一般的iii类基础油之外,润滑剂制造商还需要非常高质量的低黏度基础油,诸如通过气制油(gas
‑
to
‑
liquid)方法产生的基础油或pao型产品。仅存在少数商业气制油工厂,并且这些工厂中的大多数并不是设计来生产基础油的,因此由气制油工厂生产的优质基础油的供应非常有限。
3.低黏度聚α
‑
烯烃(pao)是优质合成基础油,工业上使用由bf3和醇组成的催化剂体系由c
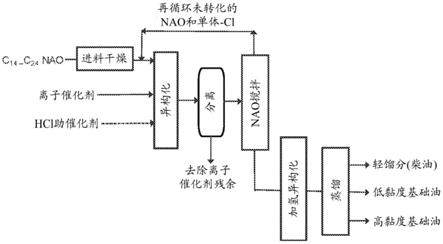
10
或c
12
正α烯烃(nao)生产。由于优秀的低温流动性质和低挥发性,低黏度pao是用于更高效率发动机的优质润滑剂的非常理想的混合原料。然而,这种pao非常昂贵,并且供应有限。因此,仍然需要用于汽车和其他应用的性质在商业可接受物理性质范围内的基础油组合物,所述物理性质包括黏度、noack挥发性和低温冷启动黏度中的一者或多者。此外,仍然需要性质有所改进的基础油组合物和制造方法。
4.许多催化方法目前用于α烯烃的低聚以生产润滑剂基本原料。
5.还已知路易斯酸(诸如bf3、alcl3和etalcl2)可作为催化剂与卤代烷(例如叔丁基氯)、醇或brdnsted酸结合用于α烯烃的阳离子聚合。
6.以引用方式并入本文的美国专利号7,527,944公开了离子液体作为催化剂用于α烯烃的阳离子聚合的用途。离子液体是过去几十年发展起来的一类化合物。本文所用的术语“离子液体”是指可通过熔化盐获得并且完全由离子组成的液体。术语“离子液体”包括具有高熔点的化合物和具有低熔点(例如等于或低于室温)的化合物两者。熔点低于大约30℃的离子液体通常称为“室温离子液体”,并且通常来源于具有含氮杂环阳离子(诸如基于咪唑鎓和吡啶鎓的阳离子)的有机盐。
7.美国专利号7,572,944公开的离子液体催化剂包含吡啶鎓或咪唑鎓阳离子以及氯铝酸根阴离子。已知离子液体作为聚合催化剂的用途提供优于常规催化剂的某些优点。特别地,离子液体一般与烃类不混溶,因此可通过相分离与聚α
‑
烯烃产物分开并再循环。相比之下,常规的路易斯酸催化剂一般在产物的分离过程中淬灭。
8.随着对高性能发动机的强烈的预期需求的增长,强烈需要并入离子催化剂来生产具有理想特性的低黏度基础油的替代方法。
技术实现要素:
9.本发明的一个实施方案是离子络合物催化剂以及用于在不存在hcl助催化剂的情况下利用离子络合物催化剂进行c
14
‑
c
24
正α烯烃的烯烃低聚的方法。
10.另外的实施方案是具有2:1无水氯化镓对1摩尔氯化铵的离子液体组合物/催化剂以及用于c
14
‑
c
24
正α烯烃的烯烃低聚的方法。
11.另外的实施方案是具有1.8:1的无水金属氯化物对1摩尔氯化铵的离子液体组合
物/催化剂以及用于c
14
‑
c
24
正α烯烃的烯烃低聚的方法。
12.另一实施方案是用于生产基础油的方法,其包括:
13.a.在存在离子催化剂的情况下,在130℃或更高的温度下,使高碳数正α烯烃反应以产生低聚物;
14.b.在h2气氛下,使用包含金属和中孔沸石的催化剂将在(a)中产生的低聚物产物加氢异构化;
15.c.蒸馏并分馏(b)的加氢异构化产物以产生最高371℃的轻馏分、在371
‑
488℃馏出物中的低黏度基础油以及高于488℃的高黏度基础油。
16.d.任选地将包含未转化的正α烯烃和有机氯化物的轻馏分再循环至反应器以通过转化步骤a。
附图说明
17.图1是由c
14
‑
c
24
nao制造优质基础油的框图。
18.图2是低聚产物的gc图,示出了c
16
氯化物、未反应的烯烃、二聚体、三聚体、四聚体和更高低聚物产物分布。
19.图3是低聚产物模拟蒸馏的曲线图。
20.图4是各种离子催化剂的低黏度选择率对c
16
nao烯烃单体转化率的曲线图。
21.图5是由c16nao产生的优质低黏度基础油合成物的支化接近度对游离碳指数、甲基支链数或支化系数的多曲线图,示出了低聚和加氢精制(hydrofinishing)步骤期间的分子结构变态。
22.图6是支化接近度对倾点或黏度指数的多曲线图,示出了分子结构变态对低黏度基础油性质的影响。
具体实施方式
23.本文描述一种使用正α烯烃来制备优质基础油的方法,所述正α烯烃具有c
14
‑
c
24
碳数范围(例如,c
14
‑
c
24 nao)、优先地c
14
‑
c
18
,所述正α烯烃可通过乙烯低聚由石油原油来源产生,或由生物基来源(诸如天然甘油三酯、脂肪酸和脂肪醇)产生,或通过蜡裂解产生。这些c
14
‑
c
24 nao将比c
10
和c
12 nao更易以更低的成本获得。此外,本文描述一种不需要hcl的使用离子络合物催化剂的方法,所述方法通过以下项提供优于现有技术的优点:(1)可使用离子络合物催化剂实现更高的nao转化率,(2)可使用如本文所述的离子络合物催化剂实现低黏度基础油馏分的更高选择率,(3)使用如本文所述的离子络合物催化剂的方法使得不期望的c
16
氯化物副产物更少,(4)离子络合物催化剂的合成成本更低。
24.基础油性质的定义
25.如本文所用,“基础油”是指用于制造包括介电流体、液压流体、压缩机流体、发动机油、润滑脂和金属加工流体的产品的油。
26.黏度是衡量基本原料流动性的物理性质。黏度是温度的强函数。两种常用的黏度量度是动态黏度和运动黏度。动态黏度衡量流体的内部流动阻力。发动机油的动态黏度量度的实例包括冷启动模拟器(ccs)黏度和微型旋转黏度计(mrv)黏度。ccs用于模拟低温启动期间曲轴轴承中的油的黏度。微型旋转黏度(mrv)衡量发动机油在于受控速率和剪切应
力下在超过45小时的时段内冷却至
‑
10℃至
‑
40℃之间的最终测试温度后的屈服应力和表观黏度。它是评估发动机油在寒冷天气下的可泵性的关键参数。动态黏度的si单位是pa s。所使用的传统单位是厘泊(cp),其等于0.001pa s(或1m pa s)。行业正在缓慢转向si单位。运动黏度是动态黏度与密度的比值。运动黏度的si单位是mm2/s。行业中其他常用的单位是40℃下的厘沲(cst)(kv40)和100℃下的厘沲(kv100)以及100℉和210℉下的赛氏通用秒(sus)。便利地,1mm2/s等于1cst。astm d5293和d445是用于测量ccs和运动黏度的相应方法。
27.黏度指数(vi)是实验数,用于衡量基本原料的运动黏度随温度的变化。vi越高,黏度随温度的相对变化越小。大多数润滑剂应用都需要高vi的基本原料,尤其是在多级汽车发动机油和其他经历大工作温度变化的汽车润滑剂中。astm d2270是普遍接受的测定vi的方法。
28.倾点是观察到试样运动时的最低温度。它是基本原料最重要的性质之一,因为大多数润滑剂被设计来在液相下工作。通常需要低倾点,尤其是在寒冷天气润滑中。astm d97是测量倾点的标准手动方法。它正逐渐被自动方法诸如astm d5950和astm d6749取代。对于本专利中的实例,使用具有1℃测试间隔的astm d5950来测量倾点。
29.挥发性是对在高温下蒸发造成的油损失的量度。由于排放和工作寿命问题,挥发性已成为非常重要的技术参数,尤其是对于较轻等级的基本原料。挥发性取决于油的分子组成,尤其是在沸点曲线的前端。noack(astm d5800)是普遍接受的测量汽车润滑剂挥发性的方法。noack测试方法本身模拟高温使用(诸如运行中的内燃机)中的蒸发损失。
30.支化系数(b1):异链烷烃中,在出现在1h nmr化学范围0.5至2.1ppm内的所有氢之中,出现在0.5至1.05ppm化学位移范围内的甲基氢的百分比。
31.支化接近度(bp):出现在13c nmr化学位移29.8ppm处的为从端基或支链上去除的四个或更多碳原子的重复亚甲基碳(ε
‑
ch2碳)的百分比。
32.每分子甲基支链数:是包括2
‑
甲基、3
‑
甲基、4
‑
甲基、5+甲基、邻甲基和出现在
13
c nmr化学位移0.5ppm和22.0ppm之间的未知甲基的数量,出现在13.8ppm处的末端甲基碳除外。
33.游离碳指数:是每分子的为从端基或支链上去除的四个或更多碳原子的亚甲基碳(ε
‑
ch2碳)的平均数。
34.13
c nmr化学位移分配:
35.支链nmr化学位移(ppm)2
‑
甲基22.53
‑
甲基19.1或11.44
‑
甲基14.05+甲基19.6内乙基10.8正丙基14.4邻甲基16.7
36.参考:assignment of the various branch carbon resonances to specific branch positions and lengths using tabulated and calculated values(lindeman,
l.p.,journal of qualitative analytical chemistry 43,1971 1245ff;netzel,d.a.等人,fuel,60,1981,307ff.)。
37.离子催化剂,如本文所述,包括离子液体催化剂、离子络合物催化剂和具有hcl助催化剂情况下的离子液体催化剂。离子液体催化剂由无水金属卤化物和季铵盐制成。alcl3、albr3、gacl3或gabr3是优选金属卤化物。烷基铵卤化物、烷基咪唑鎓卤化物和烷基吡啶鎓卤化物是优选铵盐。离子络合物催化剂由无水金属卤化物(路易斯酸)和具有强电子供体原子的分子制成,所述分子将充当路易斯碱与无水金属卤化物一起使用。alcl3、albr3、gacl3或gabr3是优选金属卤化物。尿素、硫脲、烷基酰胺和烷基膦是优选分子。
38.本文(i
‑
iii)提供制备所公开的低黏度基础油组合物的方法的实施方案。
39.i.
40.a.在存在离子催化剂的情况下,在130℃或更高的温度下,使c
14
至c
24
正α烯烃单体反应以产生低聚物;
41.b.在h2气氛下,使用包含金属和中孔沸石或二氧化硅
‑
氧化铝的催化剂将在(a)中产生的低聚物产物加氢异构化;
42.c.蒸馏和分馏(b)的加氢异构化产物以产生轻馏分(最高700℉或371℃)、700
‑
910℉(371
‑
488℃)馏出物中的低黏度基础油以及高于910℉(488℃)的高黏度基础油。
43.d.任选地将包含在步骤a中未转化的正α烯烃和有机氯化物的轻馏分再循环至反应器以通过转化步骤a。
44.上述方法的实施方案是低聚物选择率,其中低聚物产物主要是二聚体,二聚体选择率等于或大于40重量%至90%、优选地大于50%且最优选地大于60%。
45.ii.
46.a.在存在离子催化剂的情况下,在130℃或更高的温度下,使c
14
至c
24
正α烯烃单体反应以产生低聚物;
47.b.在h2气氛下,使用包含金属和中孔沸石的催化剂将在(a)中产生的低聚物产物加氢异构化;
48.c.蒸馏和分馏(b)的加氢异构化产物以产生轻馏分(最高700℉或371℃)、700
‑
910℉(371
‑
488℃)馏出物中的低黏度基础油以及高于910℉(488℃)的高黏度基础油,其中低黏度基础油表现出3.5
‑
4.6cst的100℃下的黏度,具有>130的vi、<15%的noack挥发性、<20℃的倾点和浊点以及<2500cp的
‑
35℃下的冷启动模拟器黏度的性质。
49.d.任选地将包含在步骤a中未转化的正α烯烃和有机氯化物的轻馏分再循环至反应器以通过转化步骤a。
50.iii.
51.a.在存在离子催化剂的情况下,在130℃或更高的温度下,使c
14
至c
24
正α烯烃单体反应以产生低聚物;
52.b.在h2气氛下,使用包含金属和中孔沸石的催化剂将在(a)中产生的低聚物产物加氢异构化;
53.c.蒸馏和分馏(b)的加氢异构化产物以产生轻馏分(最高700℉或371℃)、700
‑
910℉(371
‑
488℃)馏出物中的低黏度基础油以及高于910℉(488℃)的高黏度基础油,其中低黏度基础油表现出3.5
‑
4.6cst的100℃下的黏度、17.9
‑
22.2的支化接近度、20.6
‑
23.0的支
化系数、5.8
‑
7.1的游离碳指数、以及1.6
‑
2.4的每分子甲基支链数
54.d.任选地将包含在步骤a中未转化的正α烯烃和有机氯化物的轻馏分再循环至反应器以通过转化步骤a。
55.进料原料
56.进料原料由碳数范围为14至24(c
14
‑
c
24
)、优先地c
14
‑
c
18
的高碳数正α烯烃组成,所述烯烃通过石油加工产生,或由生物衍生的α烯烃产生,或通过蜡裂解产生。进料原料可包含最多40重量%的在c6‑
c
12
范围内的较低碳数正α烯烃,并且可包含最多10重量%的在c6‑
c
20
碳数范围内的石蜡。
57.烯烃低聚
58.在用于c
14
‑
c
24
低聚的方法的特定实施方案中,控制化学反应以使二聚体产率最大化并使更高分子量的低聚物(三聚体、四聚体和更高的低聚物)最小化。图1示出本发明的方法的简化框图。低聚可在合适的反应器中通过半分批模式或连续模式进行。特定实施方案是十六烯到低聚物的转化率,其中转化百分比在40%至85%的范围内。
59.在一个实施方案中,蒸馏反应混合物以去除未反应的单体。例如,可将未反应的单体与低聚物产物分离(诸如通过蒸馏),并且可将其再循环回到第一和/或第二进料原料的混合物中以用于其低聚。
60.离子催化剂
61.在本领域中已知生成离子液体催化剂的方法。以引用方式并入本文的美国专利号7,527,944公开了离子液体作为催化剂用于α烯烃的阳离子聚合的用途。离子液体是过去几十年发展起来的一类化合物。本文所用的术语“离子液体”是指可通过熔化盐获得并且完全由离子组成的液体。术语“离子液体”包括具有高熔点的化合物和具有低熔点(例如等于或低于室温)的化合物两者。熔点低于大约30℃的离子液体通常称为“室温离子液体”,并且通常来源于具有含氮杂环阳离子(诸如基于咪唑鎓和吡啶鎓的阳离子)的有机盐。
62.本文中阐述使c
14
‑
c
24
nao转化率和二聚体产率最大化的过程条件。可使用c
16 nao作为模型进料对用于c
14
‑
c
24
的二聚的各种离子液体催化剂进行测试。
63.优选离子液体组成是2摩尔无水金属氯化物对1摩尔氯化铵。这两种离子材料的混合形成完全由阳离子和阴离子制成的离子液体。
64.2mcl3+氯化铵
→
[铵阳离子]
+
[m2cl7]
‑
[0065]
其中m是选自铝、镓和铟的金属。为了改进离子液体催化剂对低黏度基础油的选择率,需要存在无水hcl助催化剂,如通过引用并入本文的us 10,435,491中报告的。
[0066]
离子液体催化剂的组成可稍微修改为1.8:1的无水金属氯化物对1摩尔氯化铵,以降低催化剂的路易斯酸性。
[0067]
优选实施方案是使用含无水氯化镓的离子液体催化剂,其与含氯化铝的催化剂相比展现出更高的对低黏度基础油的选择率。
[0068]
本发明的另一实施方案是一类用于c
14
‑
c
24
nao的烯烃低聚的离子络合物催化剂,其与常规离子液体催化剂相比展现出更高的烯烃低聚性能,具体来说,在恒定nao转化率下,离子络合物催化剂与离子液体催化剂相比提供高出约10重量%或更高的二聚体选择率。
[0069]
这类离子络合物催化剂是环境温度下的均匀熔融液体,由3:2摩尔比的无水路易
斯酸金属卤化物和路易斯碱制成。无水路易斯酸卤化物(诸如alcl3、gacl3、incl3、albr3、ali3、gabr3、gai3、inbr3和ini3)可用于制备离子络合物催化剂。合适的固体路易斯碱包括包含具有电子对的原子(诸如氧、磷、硫、氮)的分子。路易斯碱的实例包括二甲基吡啶、三甲基吡啶、烷基吡啶、三辛基膦、烷基膦、三辛基氧膦、烷基氧膦、脲类(例如,n,n'
‑
二甲基脲、n,n'
‑
二乙基脲)、硫脲类(例如,硫脲、n
‑
甲基硫脲、n,n'
‑
二甲基硫脲、n
‑
乙基硫脲、n,n'
‑
二乙基硫脲)、酰胺(例如乙酰胺、丙酰胺、苯甲酰胺)、二烷基乙酰胺、烷基酰胺、辛腈和烷基腈。当这两种固体成分(强路易斯酸和路易斯碱)以粉末形式按3:2的摩尔比混合时,它们的强相互作用导致混合的固体粉末发生深度共晶行为,然后混合物在环境温度下变为液体。
[0070]
不受任何理论的束缚,据信将强路易斯供体配体添加到第13族金属卤化物导致金属物质歧化成阳离子和阴离子的离子络合物,所述离子络合物与中性络合物处于平衡状态,诸如以下方程中所述:
[0071]
3alcl3+2l
←→
[alcl3l]+[al2cl6l]
←→
[m2cl7]
‑
+[mcl2l2]
+
[0072]
其中m是选自铝、镓和铟的金属;x是选自氯化物、溴化物和碘化物的卤化物;并且l代表路易斯碱性供体配体。共晶行为(变成液体)可来自离子物质的形成。
[0073]
加氢异构化
[0074]
作为精制步骤,在存在氢气的情况下在高压下使用加氢催化剂对低聚物产物加氢以制备完全饱和的基础油。通常使用含镍的非均相氢化催化剂进行pao加氢。也可使用贵金属(诸如pt、pd或ru)负载型催化剂进行低聚物的加氢。
[0075]
可用于本发明的加氢异构化催化剂通常包括择形分子筛、对加氢具有催化活性的金属或金属混合物以及耐高温氧化物载体。加氢组分的存在使产品稳定性得到改进。一般的催化活性加氢金属包括铬、钼、镍、钒、钴、钨、锌、铂和钯。铂和钯特别优选,铂最优选。如果使用铂和/或钯,金属含量一般在总催化剂的0.1至5重量百分比、通常0.1至2重量百分比且不超过10重量百分比的范围内。加氢异构化催化剂在例如美国专利号7,390,763和9,616,419以及美国专利申请公布2011/0192766和2017/0183583中有所讨论。
[0076]
铂和钯和钌可以是用于加氢异构化的优选金属。其他viii族过渡金属诸如ni、co、fe、w、re、os或ir可用于所述方法。可使用包含一维或二维10元环孔结构的沸石,诸如具有mfi、mel、mfs、mre、mtt、sff、sti、ton、osi或nes架构类型的那些沸石。合适的沸石包括zsm
‑
5、zsm
‑
11、zsm
‑
23、zsm
‑
35、zsm
‑
48、zsm
‑
57、ssz
‑
32、ssz
‑
35、ssz
‑
91、ssz
‑
95、ssz
‑
109、nu
‑
87、alpo
‑
31、sapo
‑
11。具有与金属结合的酸性位点的无定形材料可用于加氢异构化。合适的无定形材料包括无定形二氧化硅
‑
氧化铝、二氧化硅
‑
氧化铝
‑
二氧化钛、氧化锆
‑
氧化铝以及氧化锆
‑
氧化铈
‑
氧化铝。
[0077]
加氢异构化的条件被调整以获得具有如上所述的特定支化性质的异构化烃混合物,因此加氢异构化的条件将取决于所用进料的特性。反应温度一般在约200℃与400℃之间,优选地在260℃至370℃之间,最优选地在288℃至345℃之间,液时空速(lhsv)一般在约0.2hr
‑1与约5hr
‑1之间,优选地在约0.5hr
‑1与约3hr
‑1之间。压力一般为约15psig至约2500psig,优选地约50psig至约2000psig,更优选地约100psig至约1500psig,最优选地100psig至800psig。低压提供提高的异构化选择率,这导致进料的更多异构化和更少裂解,因此使得基本原料沸程内的烃混合物的产率增加。在加氢异构化过程期间,氢气存在于反应区中,氢气与原料的比率一般为约0.1至10mscf/bbl(千标准立方英尺/桶),优选地约0.3
至约5mscf/bbl。可将氢气与产物分离并再循环至反应区。
[0078]
蒸馏加氢异构化产物以产生三个馏分:700℉
‑
(371℃
‑
)的轻馏分、低黏度基础油馏分(700
‑
910℉或371
‑
488℃)和高黏度基础油馏分(910℉
+
或488℃
+
)。700
‑
910℉(371
‑
488℃)沸程中的低黏度馏分主要由二聚体产物构成,并且910℉
+
(488℃
+
)沸程中的高黏度馏分包含三聚体、四聚体和更高的低聚物。
[0079]
优质基础油的产物性质
[0080]
低黏度馏分主要包含二聚体和少量三聚体。高黏度馏分主要包含三聚体和四聚体。图2和图3中示出期望的低聚物产物分布的一般gc图和模拟蒸馏曲线图。
[0081]
为了满足性能要求并且满足环境要求,期望的基础油性质是低黏度、高黏度指数(vi)、低倾点和浊点、低noack挥发性和低温冷启动模拟器(ccs)黏度。
[0082]
本文所述的优质基础油性质如下(表1):
[0083]
表1
[0084]
由nao合成的优质基础油的目标性质
[0085][0086]
由高碳数正α烯烃(c
14
‑
c
24
)制成的低聚物产物具有超过150的非常高的黏度指数(vi),这远超过大于130的目标vi。然而,二聚体产物在低温性能方面具有较大缺点。低聚物产物是蜡质的,并且表现出较差的倾点和浊点低温性质。
[0087]
通过用使用含金属的中孔沸石催化剂的加氢异构化精制过程替代加氢精制步骤,基础油的性质得到进一步改进。
[0088]
在h2气氛下,使用包含贵金属和中孔沸石的催化剂将低聚产物加氢异构化,以同时使烯烃低聚物中的双键饱和并且使碳主链结构异构化。所得产物满足优质低黏度基础油的所有目标性质。不需要额外的后续加氢步骤。
[0089]
加氢异构化过程还生成若干百分率的废气和轻产物(汽油和柴油沸程烃),从而导致基础油产率的一些损失。产率损失可能高达8
‑
10体积%。非常期望将产率损失降至最低。通过在130℃或更高的高温下进行低聚以引起低聚物产物中烃主链的异构化,然后进行轻度加氢异构化,实现了这一点。通过结合这种优化的过程,实现了由起始nao生产优质基础油的超过95%的产率。
[0090]
根据本发明的低黏度基础油组合物一般将表现出以下特性:
[0091]
3.5
‑
4.6cst的100℃下的运动黏度
[0092]
17.9
‑
22.2的支化接近度
[0093]
20.6
‑
23.0的支化系数
[0094]
5.8
‑
7.1的游离碳指数
[0095]
1.6
‑
2.4的每分子甲基支链数
[0096]
130
‑
142的黏度指数
‑
20℃至
‑
60℃的倾点
[0097]
含基础油的润滑剂制剂
[0098]
本文公开的基础油可用作润滑剂基本原料来配制包含添加剂的最终润滑剂产品。在某些变型中,将根据本文所述方法制备的基本原料与一种或多种另外的基本原料共混,所述一种或多种另外的基本原料例如一种或多种可商购获得的pao、气制油(gtl)基本原料、一种或多种矿物基本原料、植物油基本原料、藻类衍生的基本原料、如本文所述的第二基本原料、或任何其他类型的可再生基本原料。可添加任何有效量的另外基本原料以获得具有期望性质的共混合基础油。例如,基于可制备的组合物的总质量,混合基础油可包括约1
‑
99%、约1
‑
80%、约1
‑
70%、约1
‑
60%、约1
‑
50%、约1
‑
40%、约1
‑
30%、约1
‑
20%或约1
‑
10%的比率的如本文所述的第一基本原料与第二基本原料(例如,可商购获得的基础油pao、gtl基本原料、一种或多种矿物基本原料、植物油基本原料、藻类衍生的基本原料、如本文所述的第二基本原料)。
[0099]
本文还公开包含本文所述的烃混合物的润滑剂组合物。在一些变型中,润滑剂组合物包含:基础油,所述基础油包含通过本文所述的任一方法产生的烃混合物的至少一部分;以及一种或多种添加剂,所述一种或多种添加剂选自由以下组成的组:抗氧化剂、黏度调节剂、降凝剂、泡沫抑制剂、去污剂、分散剂、染料、标记物、防锈剂或其他缓蚀剂、乳化剂、破乳剂、阻燃剂、抗磨剂、摩擦改进剂、热稳定性改良剂、多功能添加剂(例如,充当抗氧化剂和分散剂两者的添加剂)或它们的任何组合。润滑剂组合物可包括本文所述的烃混合物以及任何润滑剂添加剂、润滑剂添加剂的组合或可用的添加剂包。
[0100]
基于成品润滑剂组合物的总重量,用作基本原料的本文所述的任何组合物可以大于约1%存在。在某些实施方案中,基于制剂的总重量,制剂中的基本原料的量大于约2、5、15或20重量%。在一些实施方案中,基于组合物的总重量,组合物中的基础油的量为约1
‑
99%、约1
‑
80%、约1
‑
70%、约1
‑
60%、约1
‑
50%、约1
‑
40%、约1
‑
30%、约1
‑
20%或约1
‑
10%。在某些实施方案中,基于制剂的总重量,本文提供的制剂中的基本原料的量为约1%、5%、7%、10%、13%、15%、20%、30%、40%、50%、60%、70%、80%、90%或99%。
[0101]
如本领域中已知的,润滑剂添加剂的类型和量结合基础油来选择,使得成品润滑剂组合物满足特定应用的某些工业标准或规格。一般来说,基于组合物的总重量,在使用时组合物中每种添加剂的浓度的范围可为约0.001重量%至约20重量%、约0.01重量%至约10重量%、约0.1重量%至5重量%或约0.1重量%至约2.5重量%。此外,基于组合物的总重量,组合物中添加剂的总量的范围可为约0.001重量%至约50重量%、约0.01重量%至约40重量%、约0.01重量%至约30重量%、约0.01重量%至约20重量%、约0.1重量%至约10重量%或约0.1重量%至约5重量%。
[0102]
在一些变型中,本文所述的基础油被配制成润滑剂组合物以用作二冲程发动机油、变速箱油、液压油、压缩机机油、涡轮机油和润滑脂、汽车发动机油、齿轮油、船用润滑剂和过程油。过程油应用包括但不限于:轧机油、络筒油、增塑剂、锭子油、聚合物处理、脱模剂、涂料、粘合剂、密封剂、抛光剂和蜡共混物、拉伸油和冲压油、橡胶混配物、制药过程助剂、个人护理产品和油墨。
[0103]
在其他变型中,本文所述的基础油被配制作为工业油或油脂制剂,所述工业油或油脂制剂包含至少一种添加剂,所述至少一种添加剂选自抗氧化剂、抗磨剂、极压剂、消泡剂、去污剂/分散剂、防锈剂和缓蚀剂、增稠剂、增粘剂和破乳剂。还预期本发明的基本原料可配制为介电传热流体,所述介电传热流体由选自芳香烃、聚α烯烃、聚酯和天然植物油的化合物的相对纯净的共混物以及用于改进倾点、增加稳定性和降低氧化率的添加剂组成。
[0104]
将通过以下并非旨在进行限制的实施例进一步说明本发明。
[0105]
实施例1:具有无水氯化铝的离子液体催化剂
[0106]
实施例1
‑
1:n
‑
丁基吡啶鎓氯铝酸盐(c5h5nc4h9al2cl7,缩写为[bupy][al2cl7])通过以下方式合成n
‑
丁基吡啶鎓氯铝酸盐:在手套箱中在n2气氛下,将2:1摩尔比的无水alcl3粉末和干燥的n
‑
丁基吡啶鎓氯化物粉末缓慢混合在一起。在搅拌的同时轻微加热至~50℃,然后混合物变成液体。然后每次将每种固体的一小部分交替加入烧杯以继续制备熔融液体,直到所有成分都被加入并且充分溶解。继续搅拌液体过夜,然后用细小玻璃料过滤液体以去除任何残留的固体。此离子液体催化剂的组成示出在表2中。
[0107]
实施例1
‑
2:1
‑
丁基
‑3‑
甲基咪唑鎓氯铝酸盐(缩写为[bmim][al2cl7])
[0108]
使用实施例1
‑
1的程序合成此离子液体,不同的是在合成中使用1
‑
丁基
‑3‑
甲基咪唑鎓氯化物。
[0109]
实施例1
‑
3:1
‑
乙基
‑3‑
甲基咪唑鎓氯铝酸盐(缩写为[emim][al2cl7])
[0110]
使用实施例1
‑
1的程序合成此离子液体,不同的是在合成中使用1
‑
乙基
‑3‑
甲基咪唑鎓氯化物。
[0111]
实施例1
‑
4:n
‑
丁基
‑3‑
甲基吡啶鎓氯铝酸盐(c4h9n(c4h9)(ch3)al2cl7,缩写为[bmpy][al2cl7])
[0112]
使用实施例1
‑
1的程序合成此离子液体,不同的是在合成中使用n
‑
丁基
‑3‑
甲基吡啶鎓氯化物。
[0113]
实施例1
‑
5:n
‑
丁基吡啶鎓氯铝酸盐(缩写为[bupy][1.8al2cl7])
[0114]
使用实施例1
‑
1的程序和相同的起始材料合成此离子液体。然而,无水alcl3和n
‑
丁基吡啶鎓氯化物的摩尔比为1.8:1。这略微降低催化剂酸性并提高二聚体选择率。
[0115]
表2
[0116]
离子液体催化剂的组成
[0117][0118]
实施例2:具有无水氯化镓的离子液体催化剂:n
‑
丁基吡啶鎓氯镓酸盐(c5h5nc4h9ga2cl7,缩写为[bupy][ga2cl7])
[0119]
通过以下方式合成n
‑
丁基吡啶鎓氯镓酸盐:在手套箱中在n2气氛下,将2:1摩尔比的无水gacl3粉末和干燥的n
‑
丁基吡啶鎓氯化物粉末缓慢混合在一起。在搅拌的同时轻微
加热至~50℃,然后混合物变成液体。然后每次将每种固体的一小部分交替加入烧杯以继续制备熔融液体,直到所有成分都被加入并且充分溶解。继续搅拌液体过夜,然后用细小玻璃料过滤液体以去除任何残留的固体。
[0120]
实施例3:各种离子液体催化剂的烯烃低聚性能
[0121]
本实施例示出各种离子液体催化剂的性能。比较催化剂的nao低聚步骤的性能和成品加氢基础油馏分的性质。
[0122]
使用来自实施例1和2的离子液体催化剂使正十六烯正α烯烃(c
16=
nao)低聚。使用离子液体的烯烃低聚过程非常有效。在100℃下使用仅0.5体积%的离子液体催化剂分批进行。加入少量hcl助催化剂。
[0123]
用pt、pd/氧化铝催化剂对每种低聚物产物加氢。然后蒸馏最终的全部液体产物以产生三个馏分:700℉
‑
的轻馏分、低黏度基础油馏分(700
‑
910℉)和高黏度基础油馏分(910℉+),然后测量基础油性质。性能总结和最终基础油性质总结在表3中。
[0124]
表3
[0125]
用于n
‑
c
16=
nao低聚的各种离子液体催化剂的性能和加氢基础油产物的物理性质
[0126][0127]
上述结果显示,离子液体催化剂在使正α烯烃低聚方面非常活跃。反应使用仅0.5体积%的催化剂,并且实现25.5%至59.1%范围内的良好转化率。所得基础油产物具有范围是153至164的非常高的黏度指数。然而,基础油产物的低温性质较差,并且倾点和浊点远未达到目标。
[0128]
[emim][al2cl7]获得最好的冷流性质。在包含alcl3的离子液体催化剂之中,[emim][al2cl7]在低聚步骤期间提供最高的α烯烃转化率,并且产生具有更好的倾点(
‑
16℃)和浊点(
‑
14℃)同时具有略低于153的黏度指数的低黏度基础油。这表明[emim][al2cl7]催化剂在低聚期间引起碳主链的一些异构化,并使低聚物产物支化程度更高且蜡质程度更
低。这种异构化/支化降低黏度指数并改进冷性质,即降低倾点和浊点。
[0129]
含镓的离子液体催化剂[bupy][ga2cl7]对低黏度基础油更具选择性。含alcl3的离子液体催化剂(实施例3.3)和[bupy][ga2cl7](实施例3.5)表现出~47%的非常相似的烯烃转化率。[bupy][ga2cl7]催化剂表现出63.6%的低黏度油选择率,而[emim][al2cl7]表现出38.1%。
[0130]
每种催化剂的低黏度油选择率在图4中绘制为nao转化率的函数。此图比较每类离子催化剂选择性生产低黏度油的潜力。所有含氯化铝的离子液体催化剂实现对低黏度油馏分的仅约40重量%的选择率,而含氯化镓的离子液体催化剂表现出约60+重量%的选择率,提高了约20+重量%。
[0131]
实施例4
‑
1:使用离子液体催化剂的烯烃低聚过程的改进
[0132]
本实施例示出用于在低聚过程期间并入更多异构化的低聚过程改进。令人惊讶的是,发现了新的烯烃低聚条件,其提供:(1)烯烃的高转化率,(2)二聚体产物的高选择率,以及(3)成品基础油的改进很大的产物性质。性能特征的影响总结在表4中。
[0133]
表4
[0134]
离子液体催化的n
‑
c
16=
nao低聚的过程改进和加氢基础油产物的物理性质
[0135]
[0136][0137]
*:表3中的实施例4
‑
1的产物性质值来自实施例3
‑
1的分批低聚运行样品。
[0138]
使用来自实施例1
‑
5的[bupy][1.8al2cl7]对正十六碳烯正α烯烃(c
16=
nao)进行低聚。使用0.2体积%的离子液体催化剂在连续单元中进行低聚。加入少量hcl助催化剂。执行的低聚温度低至80℃,然后升至130℃和180℃。
[0139]
随着低聚的温度从80℃增加到130℃,烯烃转化率从53%下降到41%。在使用c
10 nao低聚的早期研究(us 10,435,491)中观察到这种在更高温度下的转化率下降。基于此,曾经将c
10 nao低聚的上限温度范围限制为130℃。
[0140]
当将低聚温度升高到180℃时,令人惊讶的是,烯烃转化率增加到甚至高于80℃烯烃低聚的值(在80℃下为53%,在130℃下为41%,然后在180℃下为56%)。在更高温度下,二聚体选择率也显著增加。二聚体选择率在180℃下为57%,而在80℃下仅为15%。
[0141]
在250℃、800psig下,以分批模式使用30g pt、pd/氧化铝催化剂对低聚物产物(300g)加氢6小时。然后,蒸馏最终的全部液体产物以产生三个馏分:700℉
‑
(371℃
‑
)的轻馏分、低黏度基础油馏分(700
‑
910℉或371
‑
488℃)和高黏度基础油馏分(910℉
+
或488℃
+
),然后测量基础油性质。
[0142]
表4中的结果显示,180℃的高温下的低聚显著改进成品基础油的低温性质,同时在期望范围内降低黏度指数。结果显示,130℃的低聚温度不够高到足以改进基础油的低温性质。在180℃低聚下,成品表现出
‑
20℃的倾点和
‑
5℃的浊点。这些改进使成品基础油更接近于满足表1中所示的优质基础油的目标性质,但并没有完全满足目标。
[0143]
实施例4
‑
2:对通过使用离子液体催化剂的改进烯烃低聚过程产生的基础油的结构分析
[0144]
使用核磁共振光谱法(nmr)对来自实施例4
‑
2和4
‑
3的加氢低黏度基础油产物的组成进行分析,以确定低聚过程期间碳主链的异构化和支化(甲基迁移)程度。
[0145]
使用bruker 500光谱仪获得nmr光谱。将每个样品与cdcl3以1:1(重量:重量)混
合。分析1h nmr和
13
c nmr光谱以获得结构参数。
[0146]
表5
[0147]
低聚过程变量与低黏度基础油(加氢n
‑
c
16=
nao低聚物)平均分子结构之间的关系
[0148][0149][0150]
支化接近度:总碳之中的%ε
‑
ch2碳
[0151]
支化系数:总脂肪族氢中的%甲基氢
[0152]
每分子甲基支化:分子中内部甲基的数量,不包括分子末端的伯碳甲基
[0153]
游离碳指数:每分子ε
‑
ch2碳的总数
[0154]
表5中的结果显示,180℃下的低聚增大支化系数,即在分子中造成更多甲基支化。这降低黏度指数并降低倾点和浊点。
[0155]
实施例5:各种离子络合物催化剂的合成
[0156]
实施例5
‑
1:离子络合物催化剂—topo
‑
alcl3—的合成
[0157]
通过使用200.3g无水alcl3和386.7g三辛基氧膦((c8h
17
c)3po)来制备由3:2摩尔比的无水氯化铝和三辛基氧膦制成的离子络合物。将原样的三辛基氧膦在真空烘箱中在40℃下干燥过夜。按原样使用无水氯化铝。在手套箱中进行合成。将约1/20的量的氯化铝和三辛基氧膦粉末在烧杯中用磁力搅拌器进行混合。在搅拌的同时轻微加热至~50℃,然后混合物变成液体。然后每次将每种固体的约1/20部分交替加入烧杯以继续制备熔融液体,直到所有成分都被加入并且充分溶解。继续搅拌液体过夜,然后用细小玻璃料过滤液体以去除任何残留的固体。表6中示出此离子络合物催化剂的组成。
[0158]
实施例5
‑
2:离子络合物催化剂—尿素
‑
alcl3—的合成
[0159]
通过使用480.6g无水alcl3和144.1g尿素(h2nconh2)来制备由3:2摩尔比的无水氯化铝和尿素制成的离子络合物。将原样的尿素在真空烘箱中在80℃下干燥过夜。按原样使用无水氯化铝。在手套箱中进行合成。将约1/20的量的氯化铝和尿素粉末在烧杯中用磁力搅拌器进行混合。在搅拌的同时轻微加热至~50℃,然后混合物变成液体。然后每次将每种固体的约1/20部分交替加入烧杯以继续制备熔融液体,直到所有成分都被加入并且充分溶解。继续搅拌液体过夜,然后用细小玻璃料过滤液体以去除任何残留的固体。表6中示出此离子络合物催化剂的组成。
[0160]
实施例5
‑
3:离子配位络合物—乙酰胺
‑
alcl3—的合成
[0161]
通过使用413.9g无水alcl3和122.1g乙酰胺(ch3conh2)来制备由3:2摩尔比的无水
氯化铝和乙酰胺制成的离子络合物。将原样的乙酰胺在真空烘箱中在70℃下干燥过夜。按原样使用无水氯化铝。在手套箱中进行合成。将约1/20的量的氯化铝和乙酰胺粉末在烧杯中用磁力搅拌器进行混合。在搅拌的同时轻微加热至~50℃,然后混合物变成液体。然后每次将每种固体的约1/20部分交替加入烧杯以继续制备熔融液体,直到所有成分都被加入并且充分溶解。继续搅拌液体过夜,然后用细小玻璃料过滤液体以去除任何残留的固体。表6中示出此离子络合物催化剂的组成。
[0162]
实施例5
‑
4:离子配位络合物—top
‑
alcl3—的合成
[0163]
通过使用160.2g无水alcl3和296.5g三辛基膦((c8h
17
c)3p)来制备由3:2摩尔比的无水氯化铝和三辛基膦制成的离子络合物。原样使用原样样品。在手套箱中进行合成。将约1/20的量的氯化铝粉末和三辛基膦液体在烧杯中用磁力搅拌器进行混合。在搅拌的同时轻微加热至~50℃,然后混合物变成液体。然后每次将每种组分的1/20部分交替加入烧杯以继续制备熔融液体,直到所有成分都被加入并且充分溶解。继续搅拌液体过夜,然后用细小玻璃料过滤液体以去除任何残留的固体。表6中示出此离子络合物催化剂的组成。
[0164]
表6
[0165]
离子配位络合物样品的组成
[0166][0167]
实施例6:使用alcl3和路易斯碱的离子配位络合物的c
16=
nao的烯烃低聚
[0168]
本实施例报告实施例5中描述的离子配位络合物催化剂的性能,并将结果与离子液体催化剂(实施例1.1)的结果进行比较。
[0169]
在存在实施例5的离子配位络合物催化剂的情况下使1
‑
十六烯低聚。准备配备有磁力搅拌棒、滴液漏斗和回流冷凝器的三颈2l圆底烧瓶。将约500cc的1
‑
十六烯装入烧瓶,并施加非常少量的干燥氮气吹扫,同时将液体加热至150℃。一旦圆底烧瓶中的1
‑
十六烯达到热平衡,就使用滴液漏斗以10分钟的间隔逐滴加入0.25体积%或0.5体积%的来自实施例5的离子配位络合物。加入后,低聚反应持续30多分钟以产生反应混合物。
[0170]
回收烃产物并用gc模拟蒸馏法对其进行分析,以计算c
16=
nao的转化率和对c
16
有机氯化物、低黏度基础油和高黏度基础油的选择率。结果总结在表7中。
[0171]
对使用topo
‑
alcl3催化剂的低聚物产物加氢以产生成品基础油馏分,并且性质报告在表1中。
[0172]
表7
[0173]
用于n
‑
c
16=
nao低聚的各种离子络合物催化剂的性质离子络合物催化剂与离子液体催化剂的比较
[0174]
令人惊讶的是,发现离子配位络合物催化剂比离子液体催化剂更具活性。在150℃的反应温度下,离子络合催化剂实现56.9%至81.2%的1
‑
十六烯转化率,而[bupy][alcl3]离子液体催化剂需要180℃的反应温度才实现56.1%的转化率(实施例6
‑
1至6
‑
3对比实施例3
‑
4)。
[0175]
还发现,与离子液体催化剂相比,离子配位络合物催化剂对低黏度馏分更具选择性。实施例6
‑
1对比3
‑
4具有相似的1
‑
十六烯转化率。离子络合物催化剂(实施例6.1)表现出68.2%的二聚体选择率,这优于离子液体催化剂的二聚体选择率(实施例3.4中的56.8%的二聚体选择率)。
[0176]
乙酰胺
‑
alcl3催化剂用仅0.25体积%的催化剂装载量就实现了69.2%的非常好的转化率。此催化剂表现出64重量%的非常好的二聚体选择率。
[0177]
图4还包含作为nao转化率函数的离子络合物催化剂的低黏度油选择率数据。此图比较每类离子催化剂生产低黏度油的潜力。此图显示,使用这些离子络合物催化剂可实现高达80重量%的低黏度油选择率。这是相对于包含氯化铝的基准离子液体催化剂(约40%)或含氯化镓的离子液体催化剂(约10%)的显著改进。
[0178]
另一个惊喜是不需要向催化剂添加无水hcl即可获得良好的选择率。不添加hcl,在离子络合物催化剂的情况下形成的c
16
氯化物更少。这将使产物精制更容易。
[0179]
实施例7—通过加氢异构化精制步骤对成品油性质的改进
[0180]
本实施例显示加氢异构化精制步骤用于改进优质基础油的产物性质的用途。
[0181]
使用来自实施例1
‑
1的离子液体催化剂使正十六烯正α烯烃(c
16
=nao)低聚。在100℃下使用仅0.5体积%的离子液体催化剂分批进行。加入少量hcl助催化剂。
[0182]
将低聚物产物分成三份。用pt、pd/氧化铝催化剂对一批低聚物产物加氢。使用贵金属和中孔沸石将另外两批低聚物产物加氢异构化,以同时使双键饱和并且使碳主链结构异构化。然后蒸馏最终的全部液体产物以产生三个馏分:700℉
‑
(371℃
‑
)的轻馏分、低黏度基础油馏分(700
‑
910℉或371
‑
488℃)和高黏度基础油馏分(910℉
+
或488℃
+
)。基础油馏分具有表8所示的物理性质。
[0183]
表8
[0184]
通过加氢异构化精制步骤对基础油性质的改进通过c
16=
nao低聚和精制制成的基础油的物理性质加氢与加氢异构化精制步骤的比较
[0185]
实施例7
‑
1示出通过使用离子液体催化剂进行烯烃低聚、然后使用pt,pd/al2o3催化剂加氢制成的基础油的性质。所述产物(低黏度基础油馏分和高黏度基础油馏分)分别具有152和156的优秀黏度指数。
[0186]
然而,基础油为蜡质并且冷流性质较差,因为低黏度基础油馏分表现出
‑
4℃的倾点和6℃的浊点,并且重油馏分表现出
‑
1℃的倾点和0℃的浊点。低黏度基础油馏分在较低温度下变得太蜡质,并且表现出非常差的
‑
30℃下的ccs黏度。由于冷流动性质较差,此产物不足以作为优质低黏度基础油。
[0187]
实施例7
‑
2是通过与实施例7
‑
1完全相同的低聚过程产生的基础油,不同的是进行了加氢异构化完成步骤。基础油用铂/中孔沸石/氧化铝催化剂进行精制。低黏度基础油和高黏度基础油馏分都表现出改进很大的倾点和浊点。低黏度馏分基础油具有优秀的noack挥发性、vi和良好的低至
‑
30℃下的ccs。
[0188]
实施例7
‑
3是通过与实施例7
‑
1和7
‑
2完全相同的低聚过程产生的基础油,不同的是使用另一种pt/中孔沸石/氧化铝催化剂进行了加氢异构化精制步骤。低黏度基础油和高黏度基础油馏分都表现出改进很大的倾点和浊点。低黏度馏分基础油具有优秀的noack挥发性、vi和良好的低至
‑
30℃下的ccs。
[0189]
在表8中,还将实施例7
‑
2和实施例7
‑
3的低黏度基础油馏分与从商业来源获得的合成4cst pao(比较例)进行比较。本发明的低黏度基础油馏分表现出改进很大的倾点和浊点(尽管不完全如4cst pao一样好),同时表现出比商业4cst pao更好的noack挥发性和更高的vi。
[0190]
来自实施例7
‑
2和7
‑
3的结果表明,通过使用离子液体催化剂进行低聚(二聚),然后用金属/沸石催化剂进行加氢异构化,可由高碳数nao合成优秀的低黏度基础油。结果表明,本发明的这种低黏度基础油与一般的pao不同,并且独特地适合用于以新进料源制造优质润滑剂。
[0191]
实施例8—我们的优质低黏度基础油的分子结构分析
[0192]
使用核磁共振光谱法(nmr)对来自实施例7
‑
2的加氢或加氢异构化产物的组成分析进行分析,并将其报告于下表9中(实施例8
‑
3)。还分析在较低温度加氢异构化下精制的另外两个样品,其中异构化不够有效到足以产生具有可接受低温性质的良好低黏度基础油。将这些结果与由1
‑
癸烯(l
‑
c
10=
)nao制成的4cst pao进行比较。
[0193]
表9
[0194]
平均分子结构与低黏度基础油性质之间的关系使用离子液体催化剂由n
‑
c
16=
nao产生的低黏度基础油与4cst pao的比较
[0195]
通过在低温下进行加氢异构化来精制实施例8
‑
1和8
‑
2,并且低黏度基础油性质不完全满足表1中的目标。随着产物性质的改进(实施例8
‑
3),低聚物更加异构化,如更高的支化系数(即产生更多甲基支链)所证明。异构化还降低支化接近度并减少ε
‑
ch2碳。随着c
16=
二聚体在使用沸石催化剂的加氢异构化过程中转化,分子中的线性碳链段通过甲基迁移而异构化。
[0196]
通过c
16=
nao低聚制成的低黏度基础油与由c
10=
nao产生的4cst pao非常不同,因为本发明的方法使用长得多的烯烃分子。与由c
10=
nao产生的4cst pao(比较例)相比,本发明的基础油包含更多的εch2,即更高的支化接近度和游离碳指数(实施例8
‑
1和8
‑
2),这些促成高于pao的vi的本方法的vi。由c
10 nao制成的商业4cst pao主要包含三聚体和四聚体,本质地包含更多较短的碳链,如低支化接近度和低游离碳指数所证明,同时每分子甲基支链的数量(0.9)和由c
16 nao产生的加氢低聚物的数量大约相同。
[0197]
由于在精制步骤期间应用更广泛的加氢异构化(实施例8
‑
3),我们的低黏度基础油表现出低得多的支化接近度和游离碳指数,以及更高的支化系数和更高的每分子甲基支链数。由c
16 nao制成的实施例8
‑
3现在比由c
10 nao制成的4cst pao具有更高的每分子甲基支链数。这表明在加氢异构化期间发生了甲基迁移。
[0198]
实施例9—用于获得高产率和优越产品性能的优质低黏度基础油制造方法
[0199]
可通过结合通过高温低聚步骤和使用沸石催化剂的加氢异构化步骤实现的高转化率和选择率来进一步优化总体方法,以便产生非常高质量的基础油。
[0200]
表10中所示的实施例3
‑
5示出通过100℃低聚、然后用pt,pd/氧化铝催化剂加氢制成的基础油的性质(参考案例)。
[0201]
对实施例9
‑
1至9
‑
3进行130℃和150℃下的更高温度低聚。在实施例9
‑
1(参考案例)中,然后用pt、pd/氧化铝催化剂对低聚物加氢,而在实施例9
‑
2和9
‑
3(发明)中,对低聚物进行加氢异构化精制步骤。过程条件、产物产率和性质数据总结在表10中。
[0202]
表10
[0203]
用于获得优异产物性质的优质基础油制造方法由c
16=
nao制成的优质基础油的物
理性质
[0204][0205][0206]
如在其他实施例中所见,加氢c
16=
低聚物(实施例3
‑
5和9
‑
1)表现出优秀的黏度指数和noack挥发性,但表现出较差的低温性能(较差的倾倒点和浊点,以及较差的冷启动模拟器黏度)。使用pt,pd/氧化铝催化剂(非强酸)的加氢精制步骤并不使油裂解,并且加氢步骤的成品润滑剂的产率为约100%。
[0207]
使用沸石催化剂的加氢异构化精制步骤将一些烃分子裂解成较轻的产物(废气、汽油和柴油范围产物),并且这是不期望的,因为它降低润滑剂产率。由于加氢裂解随温度
升高而增强,因此期望在满足产品要求的同时在尽可能低的温度或尽可能温和的条件下进行加氢异构化。
[0208]
在实施例9
‑
2中,通过在288℃下进行加氢异构化精制,倾点进一步下降至
‑
47℃,但基础油产率降低至92%。在实施例9
‑
3中,通过在277℃下进行加氢异构化精制,倾点下降至
‑
38℃,基础油产率为95.9%。两个样品均表现出优秀的
‑
35℃下的冷启动模拟器(ccs)黏度(1302cp和1428cp)和
‑
40℃下的微型旋转黏度计(mrv)黏度(2,006cp和58,557cp)
[0209]
表10中的结果表明,在高温低聚之后进行温和的加氢异构化精制步骤是由高分子量nao(c
14
‑
c
24 nao)合成优质低黏度基础油同时最大化总体基础油产率和基础油产物质量的有效方式。
[0210]
实施例10—我们的优质低黏度基础油的分子结构分析
[0211]
使用核磁共振光谱法(nmr)对来自实施例9
‑
1至9
‑
3的加氢或加氢异构化产物的组成分析进行分析,并将其报告于下表11中。
[0212]
表11
[0213]
平均分子结构与低黏度基础油性质之间的关系使用离子液体催化剂由n
‑
c
16=
nao产生的低黏度油与4cst pao的比较
[0214][0215][0216]
由c
16=
nao低聚制成的低黏度基础油(实施例9
‑
1)包含更多的εch2,即更高的支化接近度和游离碳指数,这促成更高的vi但也促成非常差的低温性质。由c
10 nao制成的4cst pao主要包含三聚体和四聚体,本质地包含更多较短的碳链,如低支化接近度和低游离碳指数所证明,同时每分子甲基支链的数量(0.9)和由c
16 nao产生的加氢低聚物的数量大约相同。
[0217]
由于在精制步骤期间应用更广泛的加氢异构化(实施例9
‑
2和9
‑
3),我们的低黏度基础油表现出低得多的支化接近度和游离碳指数,以及更高的支化系数和更高的每分子甲基支链数。由c
16 nao制成的实施例9
‑
2和9
‑
3现在比由c
10 nao制成的4cst pao具有更高的每分子甲基支链(2.4和2.0对比0.9)。这再次表明甲基迁移是改进产物质量的关键机制。高
温低聚和加氢异构化两者期间均发生异构化。
[0218]
实施例11—我们的优质低黏度基础油的分子结构的组成分析
[0219]
表5、9和11中报告的全部nmr结构数据绘制在图5中,以示出使用我们的方法造成的分子结构变化的影响。由于我们改变了方法:(1)使用优选的离子催化剂在较高温度下进行低聚,然后(2)使用含金属的中孔沸石催化剂进行精制加氢异构化步骤,以由c
16 nao单体产生优质低黏度基础油,所以分子结构发生实质性变化,尤其是支化接近度,从33.4变化到18.1。
[0220]
每个结构参数(支化系数、每分子甲基支链数和游离碳指数)都相对于支化接近度绘制。图5中的结果清楚地表明,分子结构变化以一致的趋势下降,并且我们可清楚地区分出我们的发明和优选的发明区域。这些曲线图显示,由c
16 nao制成的优质低黏度基础油与由c
10
nao制成的商业4cst pao的组成完全不同。这里,我们定义了通过我们的方法制成的低黏度基础油的组成。
[0221][0222]
表5、9和11中我们的产物的黏度指数和倾点在图6中相对于支化接近度nmr结构数据绘制。图6中的结果清楚地表明,低黏度基础油性质改进与分子结构变化密切相关,并且我们可清楚地区分出我们的发明和优选的发明区域。这些曲线图显示,通过我们的方法发明由c
16 nao制成的优质低黏度基础油与由c
10 nao制成的商业4cst pao的组成完全不同。这里,我们定义了通过我们的方法制成的低黏度基础油的关键性质。
[0223]
通过我们的方法发明由c
16
nao制成的优质低黏度基础油与由c
10 nao制成的商业4cst pao的组成完全不同。这里,我们定义了通过我们的方法制成的低黏度基础油的关键性质。
[0224][0225]
c
16
二聚与由c
10
三聚体制成的商业4cst相比的主要缺点是低温性能。使用我们发明的方法,我们克服了使用c
16 nao的缺陷,并且能够产生具有优秀低温性能的优质低黏度基础油,如在优秀的倾点和浊点以及ccs和mrv黏度中观察到的。