治疗吉非替尼耐药性癌症的方法
1.本技术是申请日为2006年02月02日、申请号为201810778846.6、名称为“治疗吉非替尼耐药性癌症的方法”的发明专利申请的分案申请。
2.发明背景
3.上皮细胞癌,例如,前列腺癌、乳腺癌、结肠癌、肺癌、胰腺癌、卵巢癌、脾癌、睾丸癌、胸腺癌等,是以上皮细胞的异常加速生长为特征的疾病。此加速生长最初引起肿瘤形成。最后,也可发生转移至不同的器官部位置。虽然在多种癌症的诊断和治疗方面已取得进展,但是这些疾病仍然导致显著的死亡率。
4.肺癌是工业国家中导致癌症死亡的首要原因。开始在肺中的癌症,依据细胞在显微镜下的表现,被分为两种主要类型,非小细胞肺癌和小细胞肺癌。非小细胞肺癌(鳞片细胞癌、腺癌和大细胞癌症)扩散至其它器官一般比小细胞肺癌慢。约75%的肺癌病例被归类为非小细胞肺癌(如,腺癌),而其它25%的肺癌病例为小细胞肺癌。非小细胞肺癌(nsclc)为美国、日本和西欧癌症致死的主要原因。对于晚期疾病的患者而言,化学疗法在生存率方面提供适度的帮助,但是付出显著毒性的代价,强烈需要特异性地靶向涉及肿瘤生长的关键基因损伤的治疗剂(schiller jh等,n engl j med,346:92-98,2002)。
5.表皮生长因子受体(egfr)为在上皮细胞表面表达的170千道尔顿(kda)膜结合蛋白质。egfr为许多蛋白质酪氨酸激酶(一组细胞周期调节分子)的生长因子受体家族。(w.j.gullick等,1986,cancer res.,46:285-292)。当egfr的配体(或者egf或者tgf-α)结合至胞外域时,使egfr活化,导致受体的细胞内酪氨酸激酶结构域自动磷酸化(s.cohen等,1980,j.biol.chem.,255:4834-4842;a.b.schreiber等,1983,j.biol.chem.,258:846-853)。
6.egfr为生长促进致癌基因,erbb或erbbl的蛋白质产物,其为家族即原癌基因(protooncogenes)的erbb家族的一个成员,相信在人类许多癌症的发生和发展中起关键作用。尤其是,在乳腺癌、膀胱癌、肺癌、头癌、颈癌和胃癌以及成胶质细胞瘤中观察到egfr增强的表达。致癌基因的erbb家族编码四种结构上相关的跨膜受体,即egfr、her-2/neu(erbb2)、her-3(erbb3)和her-4(erbb4)。临床上,已有报告肿瘤中的erbb致癌基因扩增和/或受体过度表达与疾病复发和患者预后差相关,以及与疗法的响应相关。(l.harris等,1999,int.j.biol.markers,14:8-15;和j.mendelsohn和j.baselga,2000,oncogene,19:6550-6565)。
7.egfr由以下三个基本结构域组成,即胞外域(ecd,其为糖基化的(glycosylated)并且含具有两个富含半胱氨酸区域的配体-结合袋(binding pocket);短跨膜结构域和具有内在酪氨酸激酶活性的细胞内结构域。跨膜区域将配体-结合结构域与细胞内结构域连接。氨基酸和dna序列分析以及egfr的非糖基化形式的研究表明,egfr蛋白质主链质量为132kda,具有1186个氨基酸残基(a.l.ullrich等,1984,nature,307:418-425;j.downward等,1984,nature,307:521-527;c.r.carlin等,1986,moi.cell.biol.,6:257-264;和f.l.v.mayes和m.d.waterfield,1984,the embo j.,3:531-537)。
8.egf或tgf-α与egfr的结合激活信号转导通路并且导致细胞增殖。egfr分子的二
聚、构象变化和内化(internalization)起传导细胞内信号的作用,引起细胞生长调节(g.carpenter和s.cohen,1979,ann.rev.biochem.,48:193-216)。影响生长因子受体功能调节或导致受体和/或配体过度表达的遗传学改变,引起细胞增殖。另外,已确定egfr在细胞分化、细胞运动性的增强、蛋白质分泌、新血管形成、癌症细胞对化学治疗剂和放射线的入侵、转移和耐药性方面起作用。(m.-j.oh等,2000,clin.cancer res.,6:4760-4763)。
9.已经鉴定多种egfr抑制剂,包括一些已经正在进行临床试验的用于治疗各种癌症的egfr抑制剂。至于近期的概要,参见de bono,j.s.和rowinsky,e.k.(2002),"the erbb receptor family:a therapeutic target for cancer",trends in molecular medicine,8,s 19-26。
10.一批在癌症治疗中用于治疗干涉的有前景的标靶,包括her-激酶轴的成员。它们在实体上皮肿瘤例如前列腺、肺和乳腺肿瘤中经常向上调节,在成胶质细胞瘤中也向上调节。表皮生长因子受体(egfr)是her-激酶轴的一个成员,并且已经是开发多种不同的癌症疗法的选择目标。egfr酪氨酸激酶抑制剂(egfr-tkis)在这些疗法当中,因为酪氨酸残基的可逆性磷酸化为egfr通路激活所必需。换言之,egfr-tkis阻滞细胞表面受体是造成引起肿瘤细胞生长和分化的触发和/或维持细胞信号通路的原因。特别是,相信这些抑制剂干扰egfr激酶结构域,称为her-i。在更有前景的egfr-tkis中,有三个系列的化合物:喹唑啉、吡啶并嘧啶和吡咯并嘧啶。
11.在临床开发中两个更先进的化合物包括吉非替尼(gefitinib)(由astrazeneca uk ltd.开发的化合物zd 1839;可以iressa的商标名获得;以下称"iressa")和埃罗替尼(erlotinib)(由genentech,inc.和osi pharmaceuticals,inc.开发的化合物osi-774,可以tarceva的商标名获得;以下称"tarceva");两者均产生令人鼓舞的临床结果。以iressa和tarceva两者进行的常规癌症治疗包括每天分别口服给予不超过500mg化合物。在2003年5月,iressa是这些产物中第一个进入美国市场的药物,此时它被批准用于晚期非小细胞肺癌患者的治疗。
12.iressa为通过直接抑制在egfr分子上酪氨酸激酶磷酸化发挥功效的口服活性喹唑啉。其竞争三磷酸腺苷(atp)结合位点,导致her-激酶轴的抑制。iressa反应的确切机制尚不完全明白,然而,研究提示,egfr的存在是是其作用的必要的先决条件。
13.在使用这些化合物中显著的局限是,其接受者可在最初对疗法的反应之后产生对它们治疗效应的耐药,或它们可不对egfr-tkis起任何可检测到级别的反应。对于egfr-tkis的反应率在不同人种组别中各异。在低端egfr-tki反应者,在某些人群中,仅百分之10-15%的晚期非小细胞肺癌患者对egfr激酶抑制剂有反应。因此,对iressa和tarceva潜在敏感的分子学机制的更好的理解,将非常有益于对那些最有可能从这样疗法中获益的患者的靶向疗法。
14.本领域明显需要对癌症,且特别是上皮细胞癌的满意治疗,所述癌症有例如肺癌、卵巢癌、乳腺癌、脑癌、结肠癌和前列腺癌,其整合tki疗法的利益并克服由患者表现出的无应答。这样的治疗可对患者,且尤其是老年患者(他们中癌症特别常见)的健康产生极大的影响。
15.发明简述
16.本发明的发明人惊奇地发现,不可逆性egfr抑制剂在对吉非替尼和/或埃罗替尼
疗法不再响应的患者中有效治疗癌症。因此,在一个实施方案中,本发明提供治疗吉非替尼和/或埃罗替尼耐药性癌症的方法。在该实施方案中,在初始给予吉非替尼和/或埃罗替尼治疗之后的时间点监测患者癌症的发展。癌症的发展是对吉非替尼和/或埃罗替尼治疗耐药的癌症的指示,并且给予患者包含不可逆性表皮生长因子受体(egfr)抑制剂的药用组合物。
17.在优选的实施方案中,不可逆性egfr抑制剂为ekb-569、hki-272或hki-357。或者,不可逆性egfr抑制剂可为结合至egfr(seq id no:1)的半胱氨酸773的任何化合物。
18.可采用本领域技术人员熟知的方法监测癌症的发展。例如,可通过对癌症的目测(visual inspection)诸如,x-线、ct扫描或mri监测其发展。或者,可通过检测肿瘤生物标记物监测癌症的发展。
19.在一个实施方案中,在整个癌症治疗的多个时间点监测患者。例如,通过分析第二时间点癌症的发展并将此分析与第一时间点的分析比较,可监测癌症的发展。第一时间点可在吉非替尼和/或埃罗替尼初始治疗之前或之后,而第二时间点在第一点之后。癌症的增大的生长表示癌症的发展。
20.在一个实施方案中,通过分析癌症的大小监测癌症的发展。在一个实施方案中,经由采用x-线、ct扫描或mri对癌症进行目测,分析癌症的大小。在一个实施方案中,通过检测肿瘤生物标记物监测癌症的大小。
21.在一个实施方案中,癌症是上皮细胞癌。在一个实施方案中,癌症是胃肠癌、前列腺癌、卵巢癌、乳腺癌、头颈癌、食道癌、肺癌、非小细胞肺癌、神经系统癌症、肾癌、视网膜癌、皮肤癌、肝癌、胰腺癌、泌尿生殖系癌症和膀胱癌。
22.在一个实施方案中,在其它的时间点监测癌症的大小,并且其它的时间点是在第二时间点之后。
23.在一个实施方案中,较晚的时间点在之前的时间点至少2个月之后。在一个实施方案中,较晚的时间点在前面的时间点至少6个月之后。在一个实施方案中,较晚的时间点在前面的时间点之后至少10个月。在一个实施方案中,较晚的时间点在前面的时间点之后至少一年。
24.在另一个实施方案中,本发明提供治疗癌症的方法,所述方法包括给予患者包含不可逆性egfr抑制剂的药用组合物,所述患者在egfr上有突变,即在seq id.no.1的790位置(t790m)上的苏氨酸被甲硫氨酸置换。t790m突变赋予对吉非替尼和/或埃罗替尼治疗的耐药性。
25.图的简述
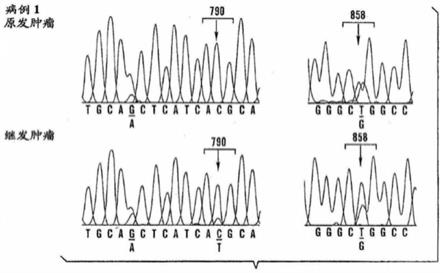
26.图1a-1b显示来自两名获得性吉非替尼耐药的nsclc患者的复发转移损伤中的egfr序列分析。图1a显示病例1的序列分析。egfr中的t790m突变存在于发展临床吉非替尼耐药之后的复发肝损伤。(左)在诊断时在原发性肺损伤中没有检测到突变。(右)原发性肺肿瘤和复发肝损伤均包含(harbor)l858r吉非替尼-敏感性突变。值得注意的是,l858r突变以期望的杂合的突变比例存在于原发性和复发损伤两者中,然而与野生型等位基因比较,t790m在低水平上是可检测的。在同样的描记图象(tracing)中显示多型性(g/a)证明非克隆pcr产物中的两个等位基因的等价物再现(seq id nos 3&4分别按出现顺序公开)。图1b显示病例2的序列分析。t790m突变存在于少数吉非替尼-耐药细胞中。(左)t790m突变或者
在肺原发性肿瘤中或者在来自该病例的八个复发肝损伤中,用测序非克隆pcr产物未能检测。在邻近多型性(g/a)的杂合性证实来自这些样本的两个egfr等位基因的扩增。杂合的吉非替尼-敏感性突变,l861q,在原发性肺肿瘤以及八个复发肝损伤中的每一个以期望的比例被检测到(seq id nos 3&5分别按出现顺序公开)。
27.图2a-2c显示支气管肺泡癌细胞系中获得对吉非替尼性耐药和对不可逆性erbb家族抑制剂的持续敏感性。图2a显示由酪氨酸激酶抑制剂对支气管肺泡癌症细胞系增殖的抑制,该细胞系具有野生型egfr(nci-hl 666)(其在egfr(nci-hl 650)中激活dele746-a750突变)或nci-hl 650(g7和cl 1)的两个代表性的吉非替尼-耐药亚克隆。可逆性抑制剂吉非替尼与不可逆性抑制剂hki-357的作用比较。用其它不可逆性抑制剂观测到可比性结果。通过在暴露于指定的药物浓度72h后,于5%fcs中和100ng/ml egfr一起培养后,经结晶紫染色,测定细胞数量。每一数据点代表四个样本的平均值。图2b显示egfr可逆性抑制剂吉非替尼、egfr不可逆性抑制剂ekb-569和两个egfr和erbb2不可逆性双重抑制剂hki-272和hki-357的化学结构。图2c显示在以不同浓度的吉非替尼或不可逆性erbb抑制剂ekb-569处理之后产生的药物-耐药性nci-hi 650细胞。在抑制剂存在下培养12天之后集落染色。
28.图3a-3d显示对吉非替尼-耐药细胞中的egfr和erbb2信号转导和改变的受体转运的持续依赖。图3a显示在带有野生型egfr(nci-hl 666)的支气管肺泡细胞系中、egfr和erbb2的sirna-介导的敲除后的细胞生存能力,与在egfr(nci-hl 650)中具有激活的dele746-a750突变和两个吉非替尼-耐药的衍生物(g7和cl 1)的细胞的生存能力进行比较。用双链rna处理72h之后计数存活的细胞,显示为用非特性sirna处理的细胞相关的分数,按一式三份样本计算标准差。图3b显示在用递增浓度的吉非替尼或不可逆性抑制剂hki-357处理、随后用egf进行2-h脉冲的细胞中,抑制下游效应器akt和mapk(erk)的egfr自动磷酸化(yl 068)和磷酸化。将亲代细胞系nci-hl 650与代表性的吉非替尼-耐药系g7比较。总的akt和mapk作为对照显示;对于在这些细胞中检测的较低限的总egfr水平,使用微管蛋白作为荷载对照。图3c显示与敏感性nci-hl 650亲代细胞系比较,在吉非替尼-耐药nci-h 1650(g7)细胞中的改变的egfr内化现象。在加入配体后5和20min时,使用若丹明标记的egf标记egfr。在nci-hl 650(g7)细胞中egfr的增强的内化现象在20min时最明显(zeiss显微镜,x 63放大倍率)。图3d显示自nci-hl 650亲代细胞内化的egfr以及经生物素化和追踪(chase)超过20min脉冲标记细胞表面蛋白之后的耐药衍生物g7的免疫印迹。将nci-hl 650(g7)细胞中的增强的细胞内egfr与不改变的铁转运蛋白受体(tr)内化现象进行比较。
29.图4a-4b显示不可逆性erbb抑制剂在抑制t790m egfr突变体中的效力。图4a显示吉非替尼和两个不可逆性抑制剂(hki-357和hki-272)抑制egfr自动磷酸化(yl 068)和nci-hl 975支气管肺泡细胞系(包含致敏性突变(l858r)和与耐药相关联的突变(t790m)两者)中的下游效应器akt和mapk(erk)的磷酸化的能力的比较。总egfr、akt和mapk显示为荷载对照。图4b显示与吉非替尼比较,用三个不可逆性erbb家族抑制剂对包含l858r和t790m突变的nci-hl 975细胞增殖的抑制。
30.图5显示egfr的核苷酸序列(seq id no:2)和氨基酸序列(seq id no:1)。
31.图6显示如同吉非替尼,hki 357和ekb 569(标识为"wyeth")被证明对包含egfr突变的nsclc细胞的增强的细胞杀伤能力,但又不同于吉非替尼,在体外不易于产生对这些药
物耐药的克隆并且它们保留其对抗吉非替尼-耐药克隆的效力。
32.发明详述
33.耐吉非替尼和埃罗替尼的癌症
34.吉非替尼(由astrazeneca uk ltd.开发的化合物zd 1839;可以iressa的商标名获得)和埃罗替尼(由genentech,inc.和osi pharmaceuticals,inc.开发的化合物osi-774,可以tarceva的商标名获得),在非小细胞肺癌(nsclcs)的病例中,产生令人惊奇的临床反应,所述癌在egf受体(egfr)(1-3)中包含激活性突变,为这些atp结合的竞争性抑制剂的标靶(4,5)。这些酪氨酸激酶抑制剂的效力既可由与这些突变相关的atp裂解(cleft)的改变(该突变导致这些药物增强对突变体激酶的抑制作用)引起,又可由这些癌症细胞对于由突变体受体转导的增强的生存信号的生物学依赖性(一种被描述为“致癌基因依赖(addiction)”的现象引起(6,7)。
35.虽然对于吉非替尼和埃罗替尼两者的治疗反应可持续长达2-3年,在大多数nsclc病例中的反应平均持续时期仅6-8个月(8-10)。获得性药物耐药的潜在的机制尚不很清楚。以此类推,用伊马替尼(imatinib)(gleevec)抑制涉及慢性髓细胞白血病(cmls)的bcr-abl激酶、牵涉胃肠道基质瘤(gists)的c-kit激酶和特发性嗜酸粒细胞增多综合征(hes)的fiplll-pdgfr-α激酶,继发性激酶结构域突变可有效地抑制药物结合(11-16)。然而,复发性nsclc不易于进行活组织检查;因此,仅有限的临床标本可用于分析。最近,报告吉非替尼或埃罗替尼疗法之后六例复发疾病的三个病例中,在egfr激酶结构域中出现单一继发性突变,t790m(17,18)。类似于egfr密码子790的bcr-abl的密码子315,在伊马替尼-耐药cml中经常突变(11,12),和c-xjr(密码子670)和fiplll-pdgfr-a(密码子674)中相应的残基突变分别与伊马替尼-耐药的gist和hes相关(15,16)。早期体外对egfr抑制剂耐药的模型表明,野生型受体中的密码子790突变将类似地抑制由egfr酪氨酸激酶抑制剂的抑制作用(19)。最近,含激活性突变的转染的egfr蛋白与t790m取代作用一起显示降低由吉非替尼和埃罗替尼的抑制作用(17,18)。虽然t790m突变似乎对在一些nsclc病例中的获得性耐药产生作用,但是其在缺乏继发性egfr突变的病例中的治疗失败的潜在机制仍然不清楚。
36.与细胞质激酶bcr-abl对比,由膜结合egfr的信号转导涉及复杂的配体结合通路、受体与erbb2和其它家族成员的同源二聚现象(homodimerization)和杂源二聚现象(heterodimerization),随后是内化现象和配体-结合受体的再循环或泛素(ubiquitin)介导的受体降解(20)。认为在内化过程发生重要的egf-依赖信号转导,其也与在细胞内囊泡中低ph下的egfr复合体的离解相关。这样,多重因素调节由受体进行的信号转导的强度和质量,并且egfr运输的改变与egf-依赖细胞性反应的调节密切关联(20)。
37.本发明基于以下发现,即吉非替尼耐药性癌症可包括那些其中t790m egfr突变仅存在于耐药肿瘤细胞亚组和那些其中没有观察到t790m突变,但观察到增强的egfr内化现象的癌症。本发明还基于以下发现,即与受体共价交联的不可逆性egfr抑制剂在抑制伴t790m突变的癌症中有效,以及在具有可使这样的癌症对用吉非替尼和/或埃罗替尼治疗耐药的改变的egfr传输的癌症中有效。因此,本发明提供包括给予不可逆性egfr抑制剂的治疗吉非替尼和/或埃罗替尼耐药性癌症的方法。
38.治疗患者的方法
39.在一个实施方案中,本发明提供治疗吉非替尼/埃罗替尼耐药性癌症的方法。该方
法包括给予需要此种治疗的患者有效量的某些不可逆性egfr抑制剂,包括ekb-569(4-苯胺基喹啉-3-腈;greenberger等,11
th nci-eortc-aacr symposium on new drugs in cancer therapy,阿姆斯特丹,2000年11月7-10日,摘要388;wyeth)、hki-357(4-苯胺基喹啉-3-腈的衍生物;tsou等,j.med.chem.2005,48:1107-1131;wyeth)和/或hki-272(4-苯胺基喹啉-3-腈的衍生物;rabindran等,cancer res.2004,64,3958-3965;wyeth)。在一个优选的实施方案中,本发明提供包括给予需要此种治疗的患者有效量的ekb-569的方法。在一个优选的实施方案中,本发明提供包括给予需要此种治疗的患者有效量的hki-357的方法。
40.该治疗还可包括治疗的组合,包括,但不限于酪氨酸激酶抑制剂与其它的酪氨酸激酶抑制剂、化学疗法、放射疗法等的组合。
41.采用在lynch等,2004;350:2129-2139描述中的方法,可将癌症最初诊断为吉非替尼/埃罗替尼敏感性或预测为吉非替尼/埃罗替尼敏感性。通过存在于肿瘤的egfr突变,包括,例如,与750(丙氨酸)中突变结合的残基747(赖氨酸)至749(谷氨酸)的缺失、残基747(赖氨酸)至750(丙氨酸)的缺失、于残基858处的精氨酸代替亮氨酸、于残基861处的谷酰胺代替亮氨酸,可预测吉非替尼/埃罗替尼敏感性。
42.在用吉非替尼和/或埃罗替尼开始治疗后,可将癌症诊断为吉非替尼和/或埃罗替尼耐药。或者,在用这些化合物初始治疗之前,可将癌症诊断为吉非替尼和/或埃罗替尼耐药。肿瘤中的吉非替尼和/或埃罗替尼耐药性可发生在吉非替尼和/或埃罗替尼治疗之后,如,6个月或更长时间。或者,在用吉非替尼和/或埃罗替尼开始治疗不到6个月之后,可诊断肿瘤的吉非替尼和/或埃罗替尼耐药。通过在吉非替尼和/或埃罗替尼治疗期间监测肿瘤的发展,可完成吉非替尼和/或埃罗替尼耐药的诊断。通过比较开始治疗后各时间点之间的肿瘤状况,或通过比较开始治疗后时间点与吉非替尼和/或埃罗替尼初始治疗之前时间点之间的肿瘤状况,可确定肿瘤的发展。在用吉非替尼和/或埃罗替尼治疗期间,可通过目测,例如,采用放射线照相术,例如,x-线、ct扫描或技术人员已知的其它监测方法,包括癌症的心悸或监测肿瘤生物标记物水平的方法,监测肿瘤的发展。在用吉非替尼和/或埃罗替尼治疗期间的癌症的发展,表明对吉非替尼和/或埃罗替尼耐药。肿瘤生物标记物水平的提高,表明肿瘤发展。因而,在用吉非替尼和/或埃罗替尼治疗期间肿瘤生物标记物水平提高,表明对吉非替尼和/或埃罗替尼耐药。检测到新的肿瘤或检测到转移,表明肿瘤发展。肿瘤收缩停止,表明肿瘤发展。癌症的生长由例如,肿瘤尺寸增大、转移或检测到新的癌症和/或肿瘤生物标记物水平提高表示。
43.通过测试来自患者循环或其它体液的、在循环中的肿瘤细胞的与吉非替尼和/或埃罗替尼耐药相关的突变存在,可监测吉非替尼和/或埃罗替尼耐药的发生。来自患者肿瘤细胞中的与吉非替尼和/或埃罗替尼耐药相关的突变的存在,是吉非替尼和/或埃罗替尼耐药肿瘤的指示。
44.在一个实施方案中,患者的肿瘤包含指示吉非替尼和/或埃罗替尼敏感性的突变,然而它对吉非替尼和/或埃罗替尼治疗是耐药的。在一个实施方案中,患者的肿瘤包含指示吉非替尼和/或埃罗替尼敏感性的突变,和包含指示吉非替尼和/或埃罗替尼耐药的突变,如,t790m突变,即在egfr中,甲硫氨酸(methione)残基取代天然的苏氨酸残基,如增强的egfr内化现象。在一个实施方案中,患者的肿瘤不包含指示吉非替尼和/或埃罗替尼敏感性的突变和不包含指示吉非替尼和/或埃罗替尼耐药的突变,如,egfr中的t790m突变,如,增
强的egfr内化现象。
45.当与药物的给予相连时,“有效量”指对至少统计学上有显著意义的部分患者导致有益效应的量,所述效应为诸如症状的改善、治愈、减轻疾病负担、缩减肿块或细胞数量、延长生命、改善生活质量或通常由熟悉治疗特殊类型的疾病或病症的医生确认为积极的其它效应。
46.所使用活性成分的有效剂量可依具体使用的化合物、给药模式、和被治疗的病症的严重程度不同而异。技术人员注意到用于每一个患者的有效剂量可依疾病严重程度、个体遗传变异或代谢速率不同而变化。然而,通常,当以约0.5-约1000mg/kg体重的每天剂量,任选一天2-4次分剂量或以缓释形式给予本发明化合物时,得到满意的结果。计划每天总剂量约1-1000mg,可优选约2-500mg。适宜于内服的的剂型包括与药学上可接受的固体或液体载体紧密混合的约0.5-1000mg的活性化合物。可调整该给药方案以提供最佳的治疗效应。例如,可每天给予多次分剂量或可依治疗情形的紧急情况的指示按比例减少剂量。
47.给药途径可为静脉内(i.v.)、肌肉内(i.m.)、皮下(s.c)、皮内(i.d.)、腹腔(i.p.)、鞘内(i.t.)、胸膜内、子宫内、直肠、阴道、局部、肿瘤内等。通过注射或整个时间渐进输液和可通过蠕动方式传输可胃肠外给予本发明化合物。
48.可通过透过粘膜或透皮方式给药。对于透过粘膜或透皮给药,在配方中应用适当的渗透屏障的渗透剂。这样的渗透剂通常为本领域所知,并且包括,例如,用于透过粘膜给药的胆汁盐类和梭链孢酸(fusidic acid)衍生物。再有,可应用清洁剂以促进渗透。透过粘膜给药可通过鼻喷雾剂,例如,或使用栓剂。对于口服给药,将本发明化合物配制成常规的口服给药形式诸如胶囊剂、片剂和滋补剂。
49.对于局部给药,将药用组合物(激酶活性抑制剂)配制成本领域通常已知的软膏剂、药膏、凝胶剂或乳膏剂。
50.本发明的治疗用组合物,如不可逆性egfr抑制剂,例如,如通过单位剂量注射经常规静脉给药。当用于指本发明的治疗用组合物时,术语“单位剂量”指适宜作为单一剂量给患者的物理上不连续的单位,每一单位含经计算产生预想治疗效应的预定量的活性物质以及与之组合的所需的稀释剂,即载体或媒介物。
51.以与剂型相协调的方式以及以有效治疗量给予组合物。将要给予的量和次数取决于被治疗患者、利用活性成分的患者系统的容量和想要的治疗效应的程度。需要给予的活性成分的精确量取决于执业医师的判断并且根据每一个体而不同。
52.本文描述了用于实施本发明方法的治疗用组合物,如,不可逆性egfr抑制剂。可应用通常为本领域技术人员已知的任何制剂或药物传递系统,其含有适合预定用途的活性成分。对于口服、直肠、局部或胃肠外(包括吸入、皮下、腹腔、肌肉内和静脉内)给药的适宜的药学上可接受的载体为本领域技术人员已知。所述载体必须为药学上可接受的,其意思是与制剂中的其它成分适配并且对其接受者没有害处。
53.如本文所使用的,术语“药学上可接受的”、“生理学上可耐受的”及其文法上的变化,在它们指组合物、载体、稀释剂和反应剂时,交替使用并且代表该物质能够是给予的,或者对于哺乳动物不产生不想要的生理效应。
54.适宜胃肠外给药的制剂便利地包括活性化合物灭菌水溶液制剂,其优选为与接受者的血液等渗。因而,这样的制剂可便利地含蒸馏水、在蒸馏水或盐水中的5%右旋糖。有用
的制剂还包括含化合物的浓缩溶液或固体,其用适当的溶剂稀释得到上述适宜胃肠外给药的溶液剂。
55.对于肠道给药,可以不连续的单位将化合物与惰性载体混合,例如胶囊剂、扁囊剂、片剂或糖锭剂,每一个含预定量的活性化合物;如粉剂或颗粒剂;或含水的或不含水的液体混悬剂或溶液剂,如,糖浆剂、酏剂、乳剂或顿服剂(draught)。适宜载体可为淀粉或糖粉和包括滑润剂、矫味剂、粘合剂及其它相同性质的原料。
56.可任选与一种或多种助剂一起,通过压制或模制,制备片剂。通过使以自由流动形式如粉状或颗粒状存在的活性化合物,任选与助剂如粘合剂、滑润剂、惰性稀释剂、表面活性剂或分散剂混合,在适宜的机器中压制,可制备压制片剂。通过将粉状的活性化合物与任何适宜载体的混合物,在适宜的机器中模制,可制备模制片剂。
57.通过加入活性化合物至浓缩的糖,如蔗糖的水溶液中,可制备糖浆剂或混悬剂,其中还可加入任何助剂。这样的助剂可包括矫味剂、阻止糖结晶的试剂或增加任何其它成分溶解的试剂,如多元醇,例如,丙三醇或山梨醇。
58.用于直肠给药的制剂可作为伴有作为栓剂基质的常规的载体,如可可脂或witepsol s55(dynamite nobel chemical,germany的商标)的栓剂形式存在。
59.用于口服给药的制剂可含有增强剂。口服可接受的吸收增强剂包括表面活性剂诸如十二烷基硫酸钠、棕榈酰肉毒碱、聚乙二醇单十二醚-9、磷脂酰胆碱、环糊精及其衍生物;胆汁盐类诸如脱氧胆酸钠、牛磺胆酸钠、甘氨胆酸盐和夫西地酸钠;螯合剂包括edta、柠檬酸和水杨酸盐;和脂肪酸(如,油酸、十二酸、酰基肉毒碱、单和双甘油酯)。其它口服吸收的增强剂包括苯扎氯铵、苄索氯铵、chaps(3-(3-胆酰氨基丙基)-二甲铵基-1-丙磺酸盐)、big-chaps(n,n-双(3-d-葡糖酰氨基丙基)-胆胺)、三氯叔丁醇、辛苯昔醇-9、苄醇、苯酚类、甲酚和烷基醇。用于本发明的特别优选的口服吸收的增强剂是十二烷基硫酸钠。
60.或者,可以脂质体或微球(或微粒)的形式给予本化合物。制备用于向患者给药的脂质体或微球的方法为本领域技术人员熟知。美国专利号4,789,734描述了脂质体中包囊生物原料的方法,其内容通过引用结合于本文。重要的是,将所述原料溶解于水溶液中,加入适当的磷脂和脂质,如果需要再加上表面活性剂,并且需要时对原料进行透析或超声处理。已知的方法的综述由g.gregoriadis,第14章,"liposomes,"drug carriers in biology and medicine,第287-341页(academic出版社,1979)提供。
61.由聚合体或蛋白质形成的微球为本领域技术人员熟悉,并且其可被定制适合于通过胃肠道直接进入血流。或者,可将该化合物整合(incorporated),并且将微球或微球合成物植入,以用于在数天至数月的时期内缓慢释放。参见,例如,美国专利号4,906,474、4,925,673和3,625,214和jein,tips 19:155-157(1998),其内容通过引用结合于本文。
62.在一个实施方案中,可将本发明的酪氨酸激酶抑制剂配制成适合尺寸的脂质体或微粒,以在静脉给药后聚集在毛细血管床。当将脂质体或微粒放入缺血组织周围的毛细血管床时,可局部给予药物至可为最有效的位点。靶向缺血组织的适宜的脂质体一般小于约200纳米并且也典型地为单层囊泡脂质体(vesicle),如在例如baldescliweiler的题目为"liposomal targeting of ischemic tissue"的美国专利号5,593,688中所公开的,其内容通过引用结合于本文。
63.优选的微粒为从生物可降解的聚合体,诸如聚乙醇酸(polyglycolide)、聚乳酸及
其共聚物制备的微粒。本领域技术人员依据包括想要的药物释放速率和想要的剂量的各种因素可易于确定适当的载体系统。
64.在一个实施方案中,经由导管(catheter)将制剂直接给药到血管内部。可例如,通过导管的孔给药。在那些其中活性化合物具有相对长的半衰期(大约1天至1周或更长)的实施方案中,制剂可纳入生物可降解的聚合性水凝胶,诸如那些在hubbell等的美国专利号5,410,016中公开的那样。可将这些聚合性水凝胶传递至组织内腔里并且在聚合体降解时活性化合物随时间推移而释放。如果需要,聚合性水凝胶可具有包含分散在其中的活性化合物的微粒或脂质体,除非有另一个机制提供活性化合物的控制释放。
65.该制剂可便利地以单位剂型存在并且可用制药领域熟悉的任何方法制备。所有方法均包括将活性化合物与组成一种或多种助剂的载体混合的步骤。通常,通过将活性化合物与液体载体或细微粉碎的固体载体均匀地和密切地混合,然后,如果需要,将所述产物定型为想要的单位剂型,制备该制剂。
66.该制剂还可包含一个或多个应用于药物制剂领域的任选的助剂,如,稀释剂、缓冲剂、矫味剂、粘合剂、表面活性剂、增稠剂、滑润剂、助悬剂、防腐剂(包括抗氧化剂)等。
67.本方法中的化合物(即不可逆性egfr抑制剂)可以向呼吸道给药的形式如鼻吸剂或气雾剂或用于喷雾器的溶液剂或如用于吹入的细微粉末存在,其单独或与惰性载体诸如乳糖混合。在这样的情况下,活性化合物的粒子适宜具有小于50微米,优选小于10微米,更优选为2-5微米的直径。
68.一般用于鼻腔给药时,弱酸ph将是优选的。优选本发明组合物的ph为约3-5,更优选为约3.5-约3.9和最优选为3.7。通过加入适当的酸诸如盐酸实现ph的调节。
69.含溶解或分散于其中的活性成分的药用组合物的制剂,为本领域熟知的且不需限于制剂。典型地这样的组合物被制备成可注射的或者液体溶液剂或者混悬剂,然而,也可制备适宜用于溶液剂或者混悬剂的固体形式,其在使用之前溶于液体。也可使制剂乳化。
70.可将活性成分以适宜用于本文描述的治疗方法的量,与药学上可接受的并且与活性成分适配的赋形剂混合。适宜赋形剂为,例如,水、盐水、右旋糖、丙三醇、乙醇等及其组合。再有,如果需要,组合物可含少量的增强活性成分效力的辅助物质诸如润湿剂或乳化剂、ph缓冲剂等。
71.本发明不可逆性激酶抑制剂可包括其成分的药学上可接受的盐。药学上可接受的盐包括与无机酸诸如,例如,盐酸或磷酸,或诸如乙酸、酒石酸、扁桃酸这样的有机酸等形成的酸加成盐(与多肽的游离氨基形成)。与游离羧基形成的盐还可衍生自无机碱,例如,氢氧化钠、氢氧化钾、氢氧化铵、氢氧化钙或氢氧化铁和诸如异丙胺、三甲胺、2-乙基氨基乙醇、组氨酸、普鲁卡因等此类有机碱。
72.生理学上可耐受的载体为本领域所熟悉。作为实例的液体载体为不含除活性成分和水之外物质的灭菌水溶液,或含缓冲液诸如生理ph值的磷酸钠、生理盐水或两者,诸如磷酸盐-缓冲盐水的灭菌水溶液。再有,水性载体可含一种以上的缓冲盐,以及盐例如氯化钠和氯化钾、右旋糖、聚乙二醇和其它溶质。
73.液体组合物还可含除水之外的以及排斥水的液相。作为实例的这样的其它液相为丙三醇、植物油诸如棉子油和水-油乳状液。
74.定义:
75.术语“erbb 1”、“表皮生长因子受体”和“egfr”可在本文中交互使用,并且指的是如在,例如在carpenter等.ann.rev.biochem.56:881-914(1987)中公开的天然序列egfr,包括其变体(如在humphrey等.pnas(usa)87:4207-4211(1990)中的缺失突变体egfr)。erbbl指编码egfr蛋白质产物的基因。如本文所使用的,egfr蛋白已作为由erbbl基因(genbank检索号nm_005228(seq id no:2))编码的genbank检索号np_005219(seq id no.:1)公开。erbbl/egfr的核苷酸和氨基酸序列可在图5中找到。
76.如本文所使用的术语“激酶活性增加的核酸变异”指基因的核苷酸序列中的变异(即突变)导致激酶活性增强。增强的激酶活性是在核酸中变异的直接结果并且与基因所编码的蛋白质相关。
77.如本文所使用的术语“药物”或“化合物”,指给予患者以治疗或预防或控制疾病或病症的化学实体或生物制品,或化学实体或生物制品的组合。化学实体或生物制品优选,但不必是低分子量化合物,而是也可为较大的化合物,例如,核酸、氨基酸的低聚体或碳水化合物包括但不限于蛋白质、寡核苷酸、核酶、dna酶、糖蛋白、sirnas、脂蛋白、适体、其修饰体及其组合。
78.如本文所使用的,术语“有效的”和“效力”包括药理学效应和生理学上安全两者。药理学效应指在患者中产生所需生物学效应的治疗能力。生理学上安全指由给予治疗导致的在细胞、器官和/或生物水平的毒性水平或其它不利的生理学上效应(通常指副作用)。“较差效应”意指治疗导致在治疗学上显著较低水平的药理学效应和/或在治疗学上较高水平的不利的生理学上效应。
79.可采用本领域熟悉的任何多种方法,从特殊的生物样本分离核酸分子,所选择的具体分离过程与特定的生物样本相适应。例如,应用冻融(freeze-thaw)和碱裂解法,可从固体材料得到核酸分子;应用加热和碱裂解法,可从尿中得到核酸分子;以及可应用蛋白酶k提取法从血液中得到核酸(rolff,a等.pcr:clinical diagnostics and research,springer(1994)。
80.如本文所使用的,在受治疗者或患者中的“癌症”指存在具有特征性的典型致癌细胞的细胞,诸如不受控制的增殖、不死性(immortality)、具转移潜力、快速生长和增殖率和某些特征性形态学特征。在某些情况下,癌症细胞将以肿瘤形式存在,或这样的细胞可局部存在于动物体内或作为独立细胞在血流中循环。
实施例
81.化合物。本文使用的化合物,包括如在以下文献中描述的ekb-569、hki-357和hki-272:美国专利号6,002,008;greenberger等,proc.11
th nci-eortc-aacr symposium on new drugs in cancer therapy,clinical.cancer res.vol.6supplement,nov.2000,issn 1078-0432;rabindran等,cancer res.64:3958-3965(2004);holbro和hynes,ann.rev.pharm.tox.44:195-217(2004);和tejpar等,j.clin.oncol.asco annual meeting proc.vol.22,no.14s:3579(2004)。
82.复发nsclc分析和吉非替尼-耐药nci-hl 650细胞产生。征得适当同意后,将尸体解剖得到复发nsclc临床样本。分析非克隆pcr产物之后,对egfr的完整激酶结构域进行测序。对外显子20的多重克隆测序以检测密码子790。通过采用染料终止剂化学(bigdye版本
1.1,applied biosystems)的单个外显子自动测序和用双向测序的侧翼内含子(flanking intronic)序列(pcr条件按要求可得到),对吉非替尼-耐药克隆中的egfr(外显子1-28)、erbb2(外显子1-24)、pten(外显子(qxons)1-9)、kras(密码子12、13和61)和p53(外显子5-8)以及亲代nci-h1650细胞系进行突变分析。在abi3100测序仪(applied biosystems)上进行测序反应和通过采用sequence navigator和factura软件(applied biosystems)分析电泳图。
83.为产生nci-hl 650细胞的耐药亚克隆,用甲磺酸乙酯(ems;600μg/ml)处理这些细胞,使恢复72h,然后接种于20μm吉非替尼中,密度为每10-cm2盘6x104个细胞。与不可逆性抑制剂比较,这些细胞对吉非替尼相对耐药,这可通过在不同药物浓度存在下,将5x104个细胞接种于5%fcs和100ng/ml egf(sigma)的六孔盘中实现,随后在用4%甲醛固定细胞72h后,用0.1%结晶紫染色,用odyssey infrared imaging system(li-cor biosciences,lincoln,ne)对细胞群定量。对于微小干预rna(sirna)敲除实验,用靶向egfr、erbb2(两个smart库(pool)来自dharmacon,lafayette,co)的双链rna寡核苷酸转染细胞,或使用x-treme基因转染反应剂(roche applied science)进行非特异性控制(lrtlb)。72h后,细胞经结晶紫染色,并用odyssey红外线扫描仪分析。
84.免疫印迹和信号传导研究。由增加吉非替尼或不可逆性抑制剂浓度对egfr信号传导的抑制通过以下实验确定:将9x104个细胞接种于24-孔盘,将药物加入含5%fcs的培养基中15min,随后用100ng/ml egf脉冲2-h,收获溶解产物。于2x凝胶荷载缓冲液中制备溶解产物,经超声波处理,煮沸,然后用10%sds/page分离,随后通过电转移至聚偏二氟乙烯(pvdf)膜并进行免疫印迹。所用的抗体为磷酸化-egfr yl 068和磷酸化促有丝分裂原活化蛋白激酶(mapk)(cell signaling technology,beverly,ma)、磷酸化-akt(biosource international,camarillo,ca)和总egfr、mapk、akt和微管蛋白(santa cruz biotechnology)。
85.egfr内化现象分析。为了用荧光显微镜方法证明egfr的内化现象,将细胞生长在盖玻片上并与1ng/ml重组人(rh)egf(分子探针,eugene,or)一起培养多个时间间隔,之后固定于4%多聚甲醛中10min。用pbs洗涤盖玻片并用prolong gold抗衰减(antifade)反应剂(分子探针)封片(mounted)。为通过细胞表面生物素化使egfr内化现象量化,将细胞生长至融合,用环己酰胺(cyclohexamide)预处理,于冰上用磺基琥珀酰亚氨基-2-(生物素基酰氨基(biotinamido))乙基-1,3-二硫代丙酸酯(磺基(sulfo)-nhs-ss-生物素;pierce)1.5mg/ml培养1h,并用封闭缓冲液(50nm nh4cl/1mm mgcl2a)1mm cacl2在pbs中)洗涤以猝灭游离的磺基-nhs-ss-生物素,然后再用pbs洗涤数次。然后将细胞于37℃培养基中培养多个时间间隔以使生物素化的分子内化,于冰上用谷胱甘肽溶液(50mm谷胱甘肽/75mm nacl/75mm naoh/1%bsa)洗涤两次20min,从细胞表面剥离所有生物素基团,然后刮下细胞并将其溶解于用naf、正钒酸钠和蛋白酶抑制剂补充的500μm放射免疫沉淀测定(ripa)缓冲液(25mm tris-hcl,ph 7.4,用150mm nacl/0.1%sds/1%triton x-100)。将细胞提取液离心,将上清液与链霉抗生物素珠(sigma)孵育以收集生物素化的蛋白,然后通过sds/page和免疫印迹,用抗-egfr抗体(sc-03,santa cruz biotechnology)或抗转铁蛋白受体抗体(santa cruz biotechnology),对生物素化的蛋白进行分析。
86.结果和讨论
87.对吉非替尼获得性耐药的复发性肺癌的分析。在两个患者中发生复发的对吉非替尼-耐药的nsclc,其肿瘤在诊断时已经包含egfr激酶的激活的突变并且已经显示引人注目的对药物的最初临床反应(1)。在两个病例中,初始治疗后1-2年,在肝中发展的转移疾病导致患者死亡。在病例1中,尸检时得到大部肝转移的分析表明,致敏egfr突变(l858r)的持续以及存在新的获得性t790m突变(图ia)。有趣的是,非克隆的pcr产物的分析显示,最初l858r突变以与存在于所有肿瘤细胞中的杂合突变相一致的丰度存在,而在约五分之一丰度的相应的野生型等位基因见到继发性t790m突变。因而,这种与耐药相关的突变似乎仅存在于在复发肿瘤的部分细胞中。
88.病例2涉及吉非替尼疗法失败后的八个在肝中明显的复发转移。在所有这些独立的损伤中,致敏的l861q egfr突变以期望的杂合突变的比率存在。通过对来自任何这些转移的非克隆pcr产物的分析,未检测到继发性egfr突变。然而,在pcr产物亚克隆之后,发现在四个转移肿瘤分析中的两个(t790m,自损伤1测序的50个克隆中的2个和自损伤2测序的56个克隆中的1个)中,t790m突变以非常低的频率存在,但从两个其它复发转移(自损伤3测序的55个克隆中的0个和自损伤4测序的59个克隆中的0个)或原发性肿瘤(75个克隆中的0个)中未能发现(图w和表1)。总之,这些结果与之前t790m突变存在于某些但不是所有的获得性吉非替尼耐药(7个肿瘤中的3个;参见参考文献17、18和21)病例的报告一致。此外,如前所指出的(18),即使在某些具有这种耐药相关突变的病例中,似乎仅存在于小部分复发损伤中的肿瘤细胞中。这些观察提示,耐药的其它的机制涉及到没有继发性egfr突变的病例中,并且这样的机制与在其它病例中的t790m突变共同存在。
89.具对不可逆性抑制剂敏感的吉非替尼-耐药细胞系的产生。鉴于egfr-突变体nsclc的临床反应和伴这些突变(2、6、22、23)的nsclc细胞系的增强的吉非替尼-敏感性之间非常相关,以及可利用的得自复发患者的临床样本受限,本技术人建立体外吉非替尼耐药模型。本技术人在20μm吉非替尼中,或者预先暴露或者没有预先暴露在诱变剂甲磺酸乙酯下,培养具有egfr激酶(dele746-a750)的框内缺失(in-frame deletion)的支气管肺泡癌细胞系nci-hl650。与表达野生型egfr(6)的某些nsclc系比较,该细胞系表现出100倍增强的对吉非替尼的敏感性。而绝大多数这些细胞被20μm吉非替尼有效杀灭,在~10
~5
的频率易于观察到耐药的克隆,不论是否用诱变剂处理。分离出四十九个独立的耐药克隆,显示对吉非替尼的敏感性平均减低50倍(图2a)。所有这些显示,没有egfr表达变化的致敏性突变的持续性,和都没有获得性的继发性egfr突变或在erbb2、p53、kras或pten中的新突变。吉非替尼-耐药克隆证明对相关的苯胺基喹唑啉类抑制剂具有相当的耐药性。然而,引人注目的是,它们表现出对erbb家族(图2a)的三个抑制剂的持续敏感性:为egfr和erbb2(对于egfr,ic
50
值分别为92和34nm,和对于erbb2,分别为59和33nm)的双重抑制剂的hki-272(24)和hki-357(参考文献25中的化合物7f),和egfr(对于egfr,ic
50
值为39nm和对于erbb2,ic
50
值为1.3μm)选择性抑制剂的ekb-569(26)(wyeth)(图2b)。此三个药物均为不可逆性抑制剂,最可能经由与egfr催化性结构域中的cys773残基或erbb2的cys805共价键合。如同吉非替尼,与表达野生型受体的细胞比较,这些化合物证明对包含egfr突变的nsclc细胞的杀灭力增强(图2a)。然而,和吉非替尼形成对照,对抗耐药克隆易于产生,即使在高的药物浓度时,本技术人未能建立在约10μm浓度对不可逆性抑制剂耐药的细胞克隆,即使在甲磺酸乙酯的诱变之后(图2c)。
90.吉非替尼-耐药细胞依赖于egfr和erbb2表达。为了洞察吉非替尼耐药性的获得和对不可逆性抑制剂的持续敏感性的机制,本技术人首先确定是否对于它们的生存能力而言,耐药细胞系依然依赖于egfr。本技术人之前已经表明,sirna-介导的egfr的敲除在包含突变体egfrs的细胞中触发细胞凋亡,但是不在具野生型等位基因的细胞中触发细胞凋亡(6)。有显著意义的是,在用sirna靶向的egfr转染之后,亲代nci-hl 650细胞及其吉非替尼-耐药衍生物显示可比性地降低细胞生存力(图3a)。因而,吉非替尼-耐药的获得不包括下游效应器的不依赖egfr的激活。因为hki-272和hki-357靶向egfr和erbb2两者,本技术人还试验此相关受体的抑制。nci-hl 650及其吉非替尼-耐药衍生物中的erbb2的敲除还引起生存能力的丧失(图3a),提示egfr-erbb2杂二聚体在包含egfr突变的肿瘤细胞中转导基本生存信号中的作用。仅由不可逆性抑制剂所致egfr的抑制似乎足以诱导吉非替尼-耐药细胞的凋亡,如被主要靶向egfr的ekb-569的效力所证明(26)。然而,鉴于通过使用sirna靶向egfr和erbb2的潜在补充效应和靶向这些家族成员的不可逆性抑制剂的可用性,双重抑制的潜在利益得到考虑。
91.经由介导其增殖和生存通路的egfr的下游效应器,本技术人比较吉非替尼和不可逆性erbb家族抑制剂对抑制信号传导的能力。在抑制egfr自动磷酸化(在残基yl 068检测)中,和包含dele746-a750 egfr突变的亲代nci-hl 650细胞中的akt和mapk磷酸化中,hki-357的效力未吉非替尼的10倍(图3b)。在吉非替尼-耐药衍生物,nci-h1650(g7)中,吉非替尼表现出相当地降低抑制akt磷酸化的效应,一种关联吉非替尼响应的关键egfr信号传导效应器(6),相反,hki-357被证明具有持续活性(图3b)。
92.吉非替尼-耐药克隆中的改变的egfr内化现象。鉴于egfr中的继发性突变的缺乏和吉非替尼-耐药细胞对sirna-介导的egfr的抑制的持续易感性,本技术人试验在吉非替尼-耐药细胞中由可逆性和不可逆性抑制剂对egfr信号传导的分化抑制的潜在机制是否可能与受体传输的改变相关,这种传输已被充分证明为egfr-依赖信号传导调节器(20)。确实,与药物-敏感性亲代细胞比较,如采用荧光素-标记的egf内化现象(图3c)和细胞质生物素化egfr的定量)两者所检测的,nci-hl 650-衍化的耐药细胞的egfr传输的分析证明与egfr内化现象的增强一致(图3d。这样的效应没有在转铁蛋白受体中观察到,提示此不是来自在所有受体过程(receptor processing)中普遍改变的结果。尽管需要进行进一步的工作阐释egfr传输中此种改变的准确机制,但该复杂过程中已经牵涉到许多调节蛋白,这些结果提示吉非替尼抑制egfr激活的能力危及这些细胞,而不可逆性抑制剂的作用为可不可检测地受到影响。
93.由不可逆性抑制剂引起的t790m egfr信号传导的抑制和增强的细胞杀伤力。由不可逆性erbb抑制剂的对egfr信号传导增强的抑制产生这样的可能性,即这些药物也可在包含egfr中的t790m继发性突变的细胞的情况中表现持续活性。因此本技术人测试这些抑制剂对包含egfr中的l858r和t790m突变的nci-hl 975支气管肺泡癌细胞系的效应(18)。有意义的是,该细胞系源自没有用egfr抑制剂治疗的患者,表明该突变不是独特地与获得性药物抵抗相关。在抑制配体-引起的egfr自动磷酸化及其下游信号传导中,当通过akt和mapk磷酸化测定时,hki-357和hki-272均比吉非替尼明显地更为有效(图aa)。类似地,所有三个不可逆性抑制剂在该细胞系中,在其对吉非替尼耐药的条件下抑制增殖(图4b)。因而,不可逆性erbb抑制剂似乎在包含t790m egfr的细胞中以及在伴有野生型受体的改变的传输的
细胞中有效。
94.本技术人的结果确认egfr中的t790m突变作为继发性突变的报告,该突变之前发生于包含激活性突变的敏感性nsclcs中,与获得性耐药的出现相关(17,18)。然而,该突变仅存在于一个亚组病例中,即使包含t790m突变的肿瘤可仅包含一小部分具有该突变的细胞。这些观察意味着多重耐药机制可共同存在于最初对吉非替尼或类似的可逆性egfr抑制剂有反应之后的复发肿瘤中。此外,这些发现提示不依赖t790m的耐药机制,如果不比在赋予药物抵抗中的t790m取代本身更有效,也可能是相同的,并可解释为什么复发肿瘤几乎不表现对于t790m的克隆状态(clonality)(17,18)。获得性吉非替尼耐药的体外机制不显著频繁涉及继发性egfr突变,而是与改变的受体传输相关。然而,应该指出的是,本技术人没有测试在所有本技术人体外建立的耐药克隆中的egfr传输,并且可能在某些克隆中存在可对吉非替尼耐药起作用的另外的机制。虽然如此,事实上所有吉非替尼-耐药克隆表现出对不可逆性erbb抑制剂的可比较的敏感性。
95.本技术人的结果表明竞争性egfr抑制剂诸如吉非替尼(其效力受到体外耐药性快速发展的限制)与不可逆性抑制剂(对其的获得性耐药似乎很少见)之间显著不同(图2c)。本技术人推测耐药细胞中的配体-结合的egfr的增强内化现象,可与在细胞内囊泡的低ph下吉非替尼-egfr复合体的离解(dissociation)关联。与此对照,受体的不可逆性交联将不受这样的受体传输的改变的影响。在缺乏药物情况下细胞经过传代最多至20代后,对吉非替尼的获得性耐药稳定持续,提示调节egfr转换(turnover)的基因的遗传或外遗传的改变可解释此现象。因为受体传输不容易通过采用可用的临床样本进行研究,所以在可能的临床相关性之前进行这样的基因改变的鉴定可能是必须的。虽然如此,这样的机制可对egfr中不具有继发性突变的复发疾病患者体内获得性吉非替尼-耐药起作用。
96.不可逆性erbb抑制剂还似乎在克服由t790m突变介导的吉非替尼耐药性中有效,所述突变推测为不论这种重要残基的改变与否,因维护抑制剂结合所传输的效应。当在进行此工作时,另一种egfr不可逆性抑制剂[cl-387,785,calbiochem(27)]显示抑制t790m egfr突变体的激酶活性(17)。在t790m的条件下的cl-387,785的效力被认为是在苯胺基团3号位置上缺乏氯(chloride)的结果,所述基团存在于吉非替尼中并且推定在空间上阻碍对在密码子790上突变体甲硫氨酸的结合。然而,ekb-569、hki-272和hki-357均在苯胺环的该位置上具有氯部分,提示它们的共享的对egfr不可逆结合能力或许解释它们的效力,而不是缺乏与t790m的特异性空间上相互作用(24-26)。因而,除了t790m突变外,可证明这些不可逆性抑制剂在包括多种耐药机制中也是广泛有效的。
[0097]
表1.egfr t790m突变在病例2的复发肿瘤中以非常低的频率存在
[0098]
[0099]
大量克隆的pcr产物测序揭示,四例肝损伤中的两例的少数等位基因含t790m突变。
[0100]
本说明书全文中引用的参考文献通过对其全文的引用结合于本文。
[0101]
具体实施方式:
[0102][0103]
1.一种治疗吉非替尼和/或埃罗替尼耐药性癌症的方法,所述方法包括以下步骤:
[0104]
a.在起先用吉非替尼和/或埃罗替尼治疗患者后的时间点上,监测患者癌症的发展,其中所述癌症的发展为对吉非替尼和/或埃罗替尼治疗耐药的癌症的指征;和
[0105]
b.给予对吉非替尼和/或埃罗替尼治疗耐药的癌症患者含不可逆性表皮生长因子受体(egfr)抑制剂的药用组合物。
[0106]
2.实施方式1的方法,其中所述不可逆性egfr抑制剂选自ekb-569、hki-272和hki-357。
[0107]
3.实施方式1的方法,其中所述不可逆性egfr抑制剂结合于egfr的半胱氨酸773(seq id no:1)。
[0108]
4.实施方式1的方法,其中所述癌症的发展通过对癌症的目测来监测。
[0109]
5.实施方式4的方法,其中所述癌症的目测通过x-线、ct扫描或mri进行。
[0110]
6.实施方式1的方法,其中所述癌症的发展通过肿瘤生物标记物检测来监测。
[0111]
7.实施方式1的方法,其中监测癌症的发展包括将第二时间点的癌症与第一时间点的癌症对比,其中第二时间点在第一时间点之后,其中第一时间点在起先用吉非替尼和/或埃罗替尼治疗之前或之后,其中癌症的生长增加指示癌症的发展。
[0112]
8.实施方式7的方法,其中其它的时间点的癌症与之前的时间点的癌症作比较。
[0113]
9.实施方式1的方法,其中所述癌症是上皮细胞癌。
[0114]
10.实施方式1的方法,其中所述癌症是胃肠癌、前列腺癌、卵巢癌、乳腺癌、头颈癌、食道癌、肺癌、非小细胞肺癌、神经系统癌症、肾癌、视网膜癌、皮肤癌、肝癌、胰腺癌、泌尿生殖系癌症和膀胱癌。
[0115]
11.一种治疗癌症的方法,所述方法包括以下步骤:
[0116]
a.给予癌症患者吉非替尼和/或埃罗替尼;
[0117]
b.监测患者的癌症发展;和
[0118]
c.一旦表明癌症发展,择给予患者不可逆性egfr抑制剂。
[0119]
12.实施方式11的方法,其中所述不可逆性egfr抑制剂选自ekb-569、hki-272和hki-357。
[0120]
13.实施方式11的方法,其中所述不可逆性egfr抑制剂结合egfr的半胱氨酸773(seq id no:1)。
[0121]
14.实施方式11的方法,其中所述癌症的发展通过对癌症的目测来监测。
[0122]
15.实施方式14的方法,其中对癌症的目测采用x-线、ct扫描或mri进行。
[0123]
16.实施方式11的方法,其中所述癌症的发展通过肿瘤生物标记物检测来监测。
[0124]
17.实施方式11的方法,其中监测癌症的发展包括将第二时间点的癌症与第一时间点的癌症对比,其中第二时间点在第一时间点之后,其中第一时间点在起先用吉非替尼和/或埃罗替尼治疗之前或之后,其中癌症的生长增加指示癌症的发展。
receptor function.oncogene 2000;19:810-820.
[0144]
9.frederick l,wang w-y,eley g,james cd.diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas.cancer res 2000;60:1383-1387.
[0145]
10.huang h-js,nagane m.klingbeil ck,et al.the enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phophorylation and unattenuated signaling.j biol chem 1997;272:2927-2935.
[0146]
11.pegram md,konecny g,slamon dj.the molecular and cellular biology of her2/neu gene amplification/overexpression and the clinical development of herceptin(trastuzumab)therapy for breast cancer.cancer treat res 2000;103:57-75.
[0147]
12.ciardiello f,tortora g.a novel approach in the treatment of cancer targeting the epidermal growth factor receptor(一种靶向上皮生长因式受体的治疗癌症的新方法).clin cancer res.2001;7:2958-2970
[0148]
13.wakeling ae,guy sp,woodburn jr et al.zd 1839(iressa):an orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy.cancer res 2002;62:5749-5754.
[0149]
14.moulder sl,yakes fm,muthuswamy sk,bianco r,simpson jf,arteaga cl.epidermal growth factor receptor(herl)tyrosine kinase inhibitor zdl 839(iressa)inhibits her2/neu(erbb2)-overexpressing breast cancer cells in vitro and in vivo.cancer res 2001;61:8887-8895.
[0150]
15.moasser mm,basso a,averbuch sd,rosen n.the tyrosine kinase inhibitor zdl 839("iressa")inhibits her2-driven signaling and suppresses the growth of her-2overexpressing tumor cells.cancer res 2001;61:7184-7188.
[0151]
16.ranson m,hammond la,ferry d,et al.zdl 839,a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor,is well tolerated and active in patients with solid,malignant tumors:results of a phase i trial.j clin oncol.2002;20:2240-2250.
[0152]
17.herbst rs,maddox a-m,rothernberg ml,等.选择性口服表皮生长因子受体酪氨酸激酶抑制剂zdl 839一般耐受良好并且在非小细胞肺癌和其它固体肿瘤具有活性:i期试用的结果。j clin oncol.2002;20:3815-3825。
[0153]
18.baselga j,rischin jb,ranson m,et al.phase i safety,pharmacokinetic and pharmacodynamic trial of zd 1839,a selective oral epidermal growth factor receptor tyrosine kinase inhibitor,in patients with five selected solid tumor types.j clin one 2002;20:4292-4302.
[0154]
19.albanell j,rojo f,averbuch s等。表皮生长因子受体抑制剂zdl 839在癌症患者的皮肤中的药效学研究:受体抑制的组织病理学和分子学推论。j clin oncol.2001;20:110-124。
tyrosine kinome in colorectal cancers.science 2003;300:949.
[0169]
34.daley gq,van etten ra,baltimore d.induction of chronic myelogenous leukemia in mice by the p210bcr/abl gene of the philadelphia chromosome.science 1990;247:824-30.
[0170]
35.heinrich,mc,corless cl,demetri gd,等。在伴转移胃肠基质肿瘤患者中的激酶突变和伊马替尼反应。j clin oncol 2003;21:4342-4349。
[0171]
36.li b,chang c,yuan m,mckenna wg,shu hg.resistance to small molecule inhibitors of epidermal growth factor receptor in malignant gliomas.cancer res 2003;63:7443-7450.