1.本发明涉及金属吸附技术领域,特别是涉及一种抗生物污染材料的制备方法及应用。
背景技术:
2.具有高效的金属吸附能力和抗菌性能的纳米纤维材料在重金属吸附、医疗、水处理等领域具有广泛的应用,一般由聚酯、聚丙烯腈、聚氨酯等材料组成。
3.静电纺丝制备的抗生物污染材料具有高比表面积、高孔隙率以及内部连通的开孔结构等突出优势,从而使其在重金属离子的吸附分离方面表现出较好的吸附性能和循环使用性能。
4.由于使用环境中存在各种微生物,导致抗生物污染材料吸附金属离子运行一段时间后,由于严重生物淤积和微生物污染,将严重影响膜的吸附性能,偕胺肟的结构将被生物破坏,因此急需开发一种抗生物污染的膜材料。
技术实现要素:
5.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种流程简单、具有抗菌性能、循环使用性能稳定的重金属离子吸附用抗生物污染材料和其制备方法。
6.为实现上述目的及其他相关目的,本发明的一方面提供一种抗生物污染材料的制备方法,包括如下步骤:
7.1)将聚丙烯腈与有机溶剂混合成纺丝液,将所述纺丝液经静电纺丝制备获得聚丙烯腈纤维膜;
8.2)将步骤1)所述的聚丙烯腈纤维膜和聚六亚甲基胍进行接枝反应制备获得接枝聚丙烯腈纤维膜;
9.3)将步骤2)所述的接枝聚丙烯腈纤维膜与盐酸羟胺进行偕胺肟化反应后活化处理获得抗生物污染材料。
10.在本发明的一些实施方式中,所述步骤1)中,所述聚丙烯腈的数均分子量为150000~200000。
11.在本发明的一些实施方式中,所述步骤1)中纺丝液中聚丙烯腈浓度为5~20wt.%。
12.在本发明的一些实施方式中,所述步骤1)中有机溶剂选自n,n-二甲基甲酰胺和/或n-甲基吡咯烷酮;所述n,n-二甲基甲酰胺和n-甲基吡咯烷酮的体积比为1:1~1:10。
13.在本发明的一些实施方式中,所述步骤1)中,所述静电纺丝的纺丝电压为16~25kv;相对湿度30%~50%;纺丝喷头到接收器的距离为8~15cm;纺丝液的推进速度为0.002mm/s~0.025mm/s。
14.在本发明的一些实施方式中,还包括在所述步骤1)中经静电纺丝将所述纺丝液在衬底上沉积;所述衬底选自pet无纺布。
15.在本发明的一些实施方式中,所述步骤2)中聚六亚甲基胍是由盐酸胍和1,6-己二胺在110℃~140℃下反应制备获得;所述盐酸胍和己二胺的摩尔比为0.5~2;优选为0.8~1.2。
16.在本发明的一些实施方式中,所述步骤2)中聚六亚甲基胍的分子量为1100~1800。
17.在本发明的一些实施方式中,所述步骤2)中聚六亚甲基胍和接枝聚丙烯腈纤维膜的质量比为1:1~8;优选为1:5~6。
18.在本发明的一些实施方式中,所述步骤2)中聚六亚甲基胍溶于溶剂中;所述溶剂选自水、四氢呋喃、三氟乙酸、n-甲基吡咯烷酮、n,n-二甲基甲酰胺的一种或多种的组合。
19.在本发明的一些实施方式中,所述步骤2)中反应温度为110℃~160℃;优选为120℃~130℃。
20.在本发明的一些实施方式中,所述步骤2)中反应时间为12~16h。
21.在本发明的一些实施方式中,所述步骤3)中,所述盐酸羟胺与聚丙烯腈的氰基的摩尔比为1:0.002~1:0.5。
22.在本发明的一些实施方式中,所述步骤3)中,偕胺肟化反应中,通过碳酸钠和/或碳酸氢钠调节ph为7~10;所述的/或碳酸氢钠的量与盐酸羟胺的质量比为1:1~1:3。
23.在本发明的一些实施方式中,所述步骤3)中,所述偕胺肟化反应肟化结束后清洗至中性。
24.本发明另一方面提供一种抗生物污染材料,采用如本发明所述的抗生物污染材料的制备方法制备获得。
25.在本发明的一些实施方式中,所述抗生物污染材料包括聚丙烯腈纤维膜,所述聚丙烯腈纤维膜的厚度为60~200μm。
26.在本发明的一些实施方式中,所述抗生物污染材料为至少包括聚丙烯腈纤维膜和衬底层的复合膜;所述衬底层选自pet无纺布;所述pet无纺布的孔隙率为40%~80%;所述pet无纺布的厚度为30~200μm。
27.在本发明的一些实施方式中,所述聚丙烯腈纤维膜为复合膜的质量含量为5%~30%。
28.本发明另一方面提供如本发明所述的抗生物污染材料在金属离子吸附领域的应用。
29.在本发明的一些实施方式中,所述金属离子选自uo2(co3)
34-、pb
2+
、ni
2+
、co
2+
、ce
3+
、cr
6+
、au
+
、cu
2+
、k
+
、ca
2+
、mg
2+
、na
+
、fe
2+
、fe
3+
中的一种或多种的组合。
具体实施方式
30.本发明的申请人经过大量实验意外发现,可以利用聚六亚甲基胍和聚丙烯腈膜(pan)进行接枝反应,生成具有抗生物污染的新型膜材料,本发明的抗生物污染材料为纳米生物膜,用于提取贵/重金属离子,以及有机相中金属离子的脱除。在此基础上,完成了本发明。
31.本发明第一方面提供一种抗生物污染材料的制备方法,包括如下步骤:
32.1)将聚丙烯腈与有机溶剂混合成纺丝液,将所述纺丝液经静电纺丝制备获得聚丙
烯腈纳米纤维层;
33.2)将步骤1)所述的聚丙烯腈纳米纤维层和聚六亚甲基胍进行接枝反应制备获得接枝聚丙烯腈纤维膜;
34.3)将步骤2)所述的接枝聚丙烯腈纤维膜与盐酸羟胺进行偕胺肟化反应后活化处理获得抗生物污染材料。
35.本发明所提供的抗生物污染材料的制备方法中,所述步骤1)是将聚丙烯腈与有机溶剂混合成纺丝液。其中所述聚丙烯腈的数均分子量为150000~200000。在一些实施例中,所述聚丙烯腈的数均分子量也可以为150000~160000,160000~170000,170000~180000,180000~190000,或190000~200000。纺丝液中聚丙烯腈浓度为5~20wt.%。在一些实施例中,纺丝液中聚丙烯腈浓度可以为5~10wt.%,10~15wt.%,或15~20wt.%。进一步的,有机溶剂可选自n,n-二甲基甲酰胺和/或n-甲基吡咯烷酮,通常情况下,可以选择n,n-二甲基甲酰胺和n-甲基吡咯烷酮的体积比为1:1~1:10,1:1~1:2,1:2~1:4,1:4~1:6,1:6~1:8,或1:8~1:10。。
36.本发明所提供的抗生物污染材料的制备方法中,所述步骤1)还包括将所述纺丝液经静电纺丝制备获得聚丙烯腈纤维膜。静电纺丝过程中,可控制纺丝电压为16~25kv,16~20kv,或20~25kv。相对湿度控制在30%~50%,30%~40%,或40%~50%。进一步的,纺丝喷头到接收器的距离为8~15cm,纺丝液的推进速度为0.002mm/s~0.025mm/s,0.002mm/s~0.005mm/s,0.005mm/s~0.01mm/s,0.01mm/s~0.015mm/s,0.015mm/s~0.02mm/s,或0.02mm/s~0.025mm/s。
37.本发明所提供的抗生物污染材料的制备方法中,还包括在所述步骤1)中经静电纺丝将所述纺丝液在衬底上沉积;所述衬底选自pet无纺布。
38.本发明所提供的抗生物污染材料的制备方法中,步骤2)是将聚丙烯腈和聚六亚甲基胍进行接枝反应制备获得接枝聚丙烯腈纤维膜。其中聚丙烯腈的氰基会先水解成羧酸基,与聚六亚甲基胍中的氨基发生缩合反应生成酰胺基团。从而实现聚六亚甲基胍对聚丙烯腈的接枝反应,得到接枝聚丙烯腈纤维膜。
39.本发明所提供的抗生物污染材料的制备方法中,步骤2)中,聚六亚甲基胍的结构式为:
[0040][0041]
在一些实施例中,聚六亚甲基胍的分子量为1100~1800,1100~1300,1300~1500,或1500~1800。
[0042]
在一具体实施例中,聚六亚甲基胍的制备方法是将盐酸胍和1,6-己二胺在一定摩尔比下在110℃~140℃下反应1~4小时,获得高粘度的聚六亚甲基胍。更具体的,盐酸胍和己二胺的摩尔比在0.5~2之间,优选的为0.8~1.2之间,所述反应温度优选的为120~130℃,反应时间优选的为2~3小时。进一步优选地,反应后,将盐酸胍和己二胺的反应物再升温至150~160℃,维持12~24小时,以获得更佳的反应产物。
[0043]
本发明所提供的抗生物污染材料的制备方法中,需要先经聚六亚甲基胍溶解在溶剂中,所述溶剂选自水、四氢呋喃、三氟乙酸、n-甲基吡咯烷酮、n,n-二甲基甲酰胺的一种或
多种的组合。所述溶剂优选选自水、四氢呋喃、n-甲基吡咯烷酮的一种或多种的组合。
[0044]
本发明所提供的抗生物污染材料的制备方法中,步骤2)中,聚六亚甲基胍和聚丙烯腈的质量比为1:1~8。在一些实施例中,聚六亚甲基胍和聚丙烯腈的质量比可以为1:1~3,1:3~5,1:5~6,1:6~7,或1:7~8;进一步的,聚六亚甲基胍和聚丙烯腈的质量比优选为1:5~6,聚六亚甲基胍和聚丙烯腈的质量比更优选为1:7~8。
[0045]
本发明所提供的抗生物污染材料的制备方法中,步骤2)中,通常情况下,聚丙烯腈纤维膜和聚六亚甲基胍需要在一定温度下进行反应。例如反应温度可以是110℃~160℃。在一些实施例中,反应温度也可以是110℃~120℃,120℃~130℃,130℃~150℃,或150℃~160℃。进一步的,反应温度优选为120℃~130℃。通常情况下,反应时间没有特别限定,在一实施例中,反应时间为12~16h,12~14h,或14~16h。
[0046]
本发明所提供的抗生物污染材料的制备方法中,接枝反应结束后取出纤维产物,将纤维产物去离子水洗涤至ph显中性。洗涤至少3次。
[0047]
本发明所提供的抗生物污染材料的制备方法中,步骤3)是将步骤2)所述的接枝聚丙烯腈纤维膜与盐酸羟胺进行偕胺肟化反应。通常情况下,步骤2)聚丙烯腈纤维膜中聚丙烯腈的氰基不会完全反应,氰基会进一步的与盐酸羟胺肟化反应,生成偕胺肟基功能基团。
[0048]
本发明所提供的抗生物污染材料的制备方法中,步骤3)中,所述盐酸羟胺与接枝聚丙烯腈纤维膜的氰基的摩尔比为1:0.002~1:0.5。在一些实施例中,盐酸羟胺与接枝聚丙烯腈纤维膜的氰基的摩尔比为1:0.002~1:0.005,1:0.005~1:0.01,1:0.01~1:0.02,1:0.02~1:0.03,1:0.03~1:0.04,或1:0.04~1:0.05。
[0049]
本发明所提供的抗生物污染材料的制备方法中,步骤3)中,在温度为50~90℃下,将步骤2)中制备的接枝聚丙烯腈纤维膜浸没在配置好的盐酸羟胺溶液中,或将配置好的盐酸羟胺溶液通过提升泵循环打入到步骤2)中制备的接枝聚丙烯腈纤维膜中,控制盐酸羟胺溶液ph为7~10的之间,实现接枝聚丙烯腈纤维膜的偕胺肟化改性。所述ph为7~10的碱性溶液,可以通过投加碳酸钠和/或碳酸氢钠等来调节,所述的碳酸钠和/或碳酸氢钠的量与盐酸羟胺的质量比为1:1~1:3,1:1~1:2,或1:2~1:3。
[0050]
本发明所提供的抗生物污染材料的制备方法中,步骤3)中,偕胺肟化反应得到卷式抗生物污染材料。卷式抗生物污染材料浸没到含有盐酸羟胺的碱性溶液中进行偕胺肟化,肟化结束后清水清洗膜至中性,然后浸入酸性溶液中活化处理后即可获得抗生物污染材料。吸附过程完成后同样通过酸性溶液脱附后循环使用。其中,所述的碱性溶液可以使用碳酸钠、碳酸氢钠中的一种或几种;所述酸性溶液可以使用盐酸、硝酸、磷酸中的一种或几种;所述偕胺肟化的反应温度为50~90℃,50~70℃,或70~90℃。
[0051]
本发明第二方面提供一种抗生物污染材料,采用本发明第一方面所述的抗生物污染材料的制备方法制备获得。
[0052]
本发明所提供的抗生物污染材料中,所述抗生物污染材料包括聚丙烯腈纤维膜,所述聚丙烯腈纤维膜的厚度为60~200μm,60~100μm,100~150μm,或150~200μm。
[0053]
本发明所提供的抗生物污染材料中,所述抗生物污染材料还包括衬底层,聚丙烯腈纤维膜是在纺丝过程中将pan纳米纤维丝沉积在衬底层上形成。此时的抗生物污染材料可以看成至少包括聚丙烯腈纤维膜和衬底的复合膜。其中,衬底层例如可以是高分子无纺布,高分子无纺布为聚对苯二甲酸乙二醇酯(pet)材料,即pet无纺布,pet无纺布孔隙率为
40%~80%,40%~50%,50%~60%,60%~70%,或70%~80%。pet无纺布的厚度为30~200μm,30~50μm,50~80μm,80~100μm,100~150μm,或150~150μm。
[0054]
本发明所提供的抗生物污染材料中,所述聚丙烯腈纤维膜为复合膜的质量含量的5%~30%,5%~10%,10%~15%,15%~20%,20%~25%,或25%~30%。
[0055]
本发明第三方面提供本发明第二方面所述的抗生物污染材料在金属离子吸附领域的应用。金属离子例如可以是工业废水、地下水和饮用水中的其它贵/重金属离子或者有机相中金属离子。
[0056]
本发明所提供的应用中,所述金属离子选自uo2(co3)
34-、pb
2+
、ni
2+
、co
2+
、ce
3+
、cr
6+
、au
+
、cu
2+
、k
+
、ca
2+
、mg
2+
、na
+
、fe
2+
、fe
3+
等中的一种或多种的组合。
[0057]
本发明的有益效果:
[0058]
1.本发明的抗生物污染材料为纳米纤维膜,是通过电纺制备的聚丙烯腈类抗生物污染材料,工艺流程简单、工业化实施容易。
[0059]
2.本发明的抗生物污染的抗生物污染材料以聚丙烯腈,盐酸胍和己二胺为原料,原料易得。
[0060]
3.本发明的抗生物污染材料抗生物污染如革兰氏阴性大肠杆菌,革兰阳性金黄色葡萄球菌株等,和未处理的抗生物污染材料比较,处理后材料抗菌活性值可增加4.5~4.8。
[0061]
4.本发明的抗生物污染材料更加稳定,且吸附容量更高。
[0062]
5.本发明的抗污染污染材料的纳米纤维膜适用于提取贵/重金属离子,以及有机相中金属离子的脱除。
[0063]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离本发明的精神下进行各种修饰或改变。
[0064]
在下述实施例中,所使用到的试剂、材料以及仪器如没有特殊的说明,均可商购获得。
[0065]
pet无纺布购自潍坊赛洋防水材料有限公司的sy-3。聚丙烯腈购自南京邦诺生物科技有限公司,数均分子量180000。
[0066]
实施例1
[0067]
(1)将聚丙烯腈和有机溶剂混合成纺丝液,其中聚丙烯腈数均分子量为180000,聚丙烯腈浓度为10wt.%,有机溶剂选自n,n-二甲基甲酰胺(dmf)和n-甲基吡咯烷酮(nmp),dmf/nmp的体积比为1:5;通过静电纺丝制备0.1m
2 pet-pan纳米纤维膜,纺丝电压为20kv,相对湿度50%;纺丝喷头到接收器的距离为10cm,纺丝液的推进速度为0.01mm/s,制备以pet为衬底的复合的聚丙烯腈纤维膜,其中聚丙烯腈纤维含量为0.625g,聚丙烯腈纤维膜厚度为60μm。
[0068]
(2)聚六亚甲基胍的制备,将盐酸胍和1,6-己二胺等摩尔比在120℃下反应两小时,然后升至150-160℃并维持12小时,获得高粘度的聚六亚甲基胍。
[0069]
(3)将步骤(2)获得的聚六亚甲基胍0.15g,pan和聚六亚甲基胍的质量比为4:1,将聚六亚甲基胍溶解水中,加热至130℃,将步骤(1)制备的纤维膜浸泡至聚亚甲基胍水溶液中反应12小时,制备接枝聚丙烯腈纤维膜。
[0070]
(4)将步骤(3)制备的抗生物污染材料浸没到质量分数为10%的盐酸羟胺溶液中,70℃水浴震荡240min,反应过程中通过投加碳酸钠维持反应体系ph值在7-10之间,反应结束后取出纤维膜,清水反复洗涤至ph显中性,然后将肟化好的膜置于ph 0.6的盐酸溶液中处理2h后取出用清水反复洗涤ph显中性。
[0071]
对比例1
[0072]
(1)将聚丙烯腈和有机溶剂混合成纺丝液,其中聚丙烯腈数均分子量为180000,聚丙烯腈浓度为10wt.%,有机溶剂选自n,n-二甲基甲酰胺(dmf)和n-甲基吡咯烷酮(nmp),dmf/nmp的体积比为1:5;通过静电纺丝制备0.1m2pet-pan纳米纤维膜,纺丝电压为20kv,相对湿度50%;纺丝喷头到接收器的距离为10cm,纺丝液的推进速度为0.01mm/s,制备以pet为衬底的复合的聚丙烯腈纤维膜,其中聚丙烯腈纤维含量为0.625g,聚丙烯腈纤维膜层厚度为60μm。
[0073]
(2)将上述制备的抗生物污染材料浸没到质量分数为10%的盐酸羟胺溶液中,70℃水浴震荡240min,反应过程中通过投加碳酸钠维持反应体系ph值在7-10之间,反应结束后取出纤维膜,清水反复洗涤至ph显中性,然后将肟化好的膜置于ph 0.6的盐酸溶液中处理2h后取出用清水反复洗涤ph显中性。
[0074]
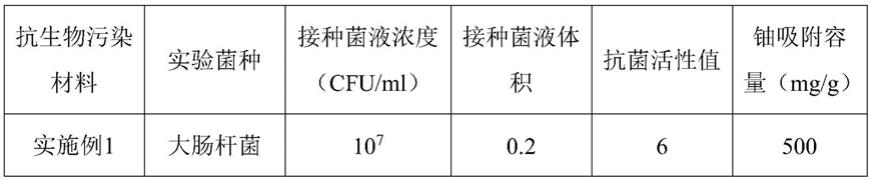
实施例1和对比例1的抗菌性能和铀吸附性能如表1
[0075]
表1
[0076][0077][0078]
实施例2
[0079]
(1)将聚丙烯腈和有机溶剂混合成纺丝液,其中聚丙烯腈数均分子量为180000,聚丙烯腈浓度为10wt.%,有机溶剂选自n,n-二甲基甲酰胺(dmf)和n-甲基吡咯烷酮(nmp),dmf/nmp的体积比为1:1;通过静电纺丝制备1m
2 pet-pan纳米纤维膜,纺丝电压为25kv,相对湿度40%;纺丝喷头到接收器的距离为8cm,纺丝液的推进速度为0.02mm/s,制备以pet为衬底的复合的聚丙烯腈纤维膜,其中聚丙烯腈纤维含量为12.5g,聚丙烯腈纤维膜厚度为60μm。
[0080]
(2)聚六亚甲基胍的制备,将盐酸胍和1,6-己二胺摩尔比在1.2:1在130℃下反应两小时,然后升至150-160℃并维持12小时,获得高粘度的聚六亚甲基胍。
[0081]
(3)将步骤(2)获得的聚六亚甲基胍1.56g,pan和聚六亚甲基胍的质量比为8:1,将聚六亚甲基胍溶解水中,加热至130℃,将步骤(1)制备的纤维膜浸泡至聚亚甲基胍水溶液中反应12小时,制备接枝聚丙烯腈纤维膜。
[0082]
(4)将上述制备的抗生物污染材料浸没到质量分数为10%的盐酸羟胺溶液中,70℃水浴震荡240min,反应过程中通过投加碳酸钠维持反应体系ph值在7-10之间,反应结束后取出纤维膜,清水反复洗涤至ph显中性,然后将肟化好的膜置于ph 0.6的盐酸溶液中处
理2h后取出用清水反复洗涤ph显中性。
[0083]
对比例2
[0084]
(1)将聚丙烯腈和有机溶剂混合成纺丝液,其中聚丙烯腈数均分子量为180000,聚丙烯腈浓度为10wt.%,有机溶剂选自n,n-二甲基甲酰胺(dmf)和n-甲基吡咯烷酮(nmp),dmf/nmp的体积比为1:1;通过静电纺丝制备1m
2 pet-pan纳米纤维膜,纺丝电压为25kv,相对湿度40%;纺丝喷头到接收器的距离为8cm,纺丝液的推进速度为0.02mm/s,制备以pet为衬底的复合的聚丙烯腈纤维膜,其中聚丙烯腈纤维含量为12.5g,聚丙烯腈纤维膜层厚度为60μm。
[0085]
(2)将上述制备的抗生物污染材料浸没到质量分数为10%的盐酸羟胺溶液中,70℃水浴震荡240min,反应过程中通过投加碳酸钠维持反应体系ph值在7-10之间,反应结束后取出纤维膜,清水反复洗涤至ph显中性,然后将肟化好的膜置于ph 0.6的盐酸溶液中处理2h后取出用清水反复洗涤ph显中性。
[0086]
实施例2和对比例2的抗菌性能和铀吸附性能如表2
[0087]
表2
[0088][0089]
前述实施例和对比例中,关于抗菌活性值和铀吸附容量测试方法如下:
[0090]
抗菌活性值的测试方法:采用jis z2801标准测试。
[0091]
铀吸附容量的测试方法:q=(c0-ct)*v/m
[0092]
其中,q是吸附容量mg/g;c0是铀的原始浓度;ct是吸附t时间后,铀的浓度v是铀溶液的体积;m是吸附剂的质量。
[0093]
上述实施例仅例示性说明本技术的原理及其功效,而非用于限制本技术。任何熟悉此技术的人士皆可在不违背本技术的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本技术所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本技术的权利要求所涵盖。