1.本发明涉及药物合成领域,具体指一种达格列净的制备方法。
背景技术:
2.达格列净,化学名:(2s,3r,4r,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)四氢-2h-呲喃-3,4,5-三醇,结构式为:
[0003][0004]
达格列净达格列净是一种钠-葡萄糖协同转运蛋白2抑制剂,美国食品药品管理局(fda)于2014年1月8日宣布,批准将达格列净用于2型糖尿病的治疗,同时要求生产商就药物相关风险开展上市后研究。
[0005]
达格列净的合成有多条路线,路线1由文献j.med.chem.2014,57,1236-1251报道,合成路线如图1;路线2由专利ep1506211b1报道的合成路线如图2;路线3由专利wo2013152476a中报道,其合成路线如图3;路线4由lemaire等人报道(org.lett.2012,14,1480-1483),其合成路线如图4;路线5由中国药物化学杂志2014,24(5),375-379报道,其合成路线如图5;路线6由专利wo 2013068850 a2报道,其合成路线如图6;路线7由lin hu et.报道(cn108530408),合成路线如图7。
[0006]
上述各反应路线可概括为图8的路线,即当r2为氢时,化合物4即为达格列净,r1为羟基保护基团或氢,r2为羰基或氢。除路线6外,其余路线的核心都是糖环与苯环的偶联(即化合物1和化合物2反应得到化合物3)以及三乙基硅烷参与的还原反应(化合物3反应得到达格列净)。在糖环与苯环偶联反应完成后产物中一般有大量的杂质存在(中间体纯度通常不超过90%),严重影响达格列净的纯度及收率。
技术实现要素:
[0007]
本发明所要解决的技术问题是针对现有技术的现状,提供一种通过新的中间体晶型化合物制备达格列净的方法,该方法能有效提高产物纯度及收率。
[0008]
本发明解决上述技术问题所采用的技术方案为:
[0009]
一种达格列净的制备方法,包括以下步骤:
[0010]
将(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇晶型a加入反应器中,加入二氯甲烷、乙腈、三乙基硅烷,降温至-20℃,然后滴加三氟化硼乙醚溶液,滴加完毕后保温反应;
[0011]
反应完毕后加入饱和碳酸钠溶液淬灭反应,所得水层经过二氯甲烷萃取后与有机层合并,然后水洗,经无水硫酸钠干燥后减压浓缩得到达格列净。
[0012]
上述(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四
氢-2h-吡喃-3,4,5-三醇的结构式为:
[0013][0014]
上述(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇的x射线粉末衍射图谱具有如下特征衍射峰:5.7
±
0.2
°
,13.1
±
0.2
°
,15.0
±
0.2
°
,18.3
±
0.2
°
,20.5
±
0.2
°
,20.8
±
0.2
°
,21.6
±
0.2
°
,23.1
±
0.2
°
和24.5
±
0.2
°
。
[0015]
上述(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇在无醇类溶剂或水参与结晶时为油状物。
[0016]
优选地,(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇晶型a中含有3.5~15%的结晶溶剂。所述的结晶溶剂为甲醇、乙醇、异丙醇、正丙醇或他们的组合,进一步优选为正丙醇。
[0017]
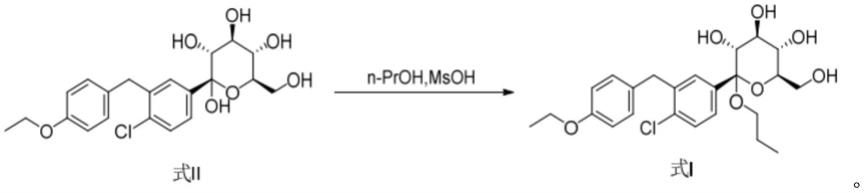
在0~30℃下,以甲磺酸为缚酸剂,式
ⅰⅰ
化合物在正丙醇溶液中反应得到式ⅰ化合物,即为(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇,
[0018][0019]
在0~30℃下,从式ⅰ化合物的正丙醇溶液中结晶式ⅰ化合物,加入相当于式ⅰ化合物质量5%~30%的水,以及在相同温度下,滴加c5~c8烷烃,结晶,得到式ⅰ化合物的晶型a。
[0020]
所述正丙醇溶液的浓度为0.1g/ml~5.0g/ml,c5~c8烷烃的加入体积为式ⅰ化合物质量的5~20倍,结晶温度为10~30℃。
[0021]
优选地,上述式ⅰ化合物的晶型a的制备方法中,分离得到的式ⅰ化合物可以为油状或无定型。可以选择在溶液中添加晶种,晶种的加入包括在将c5~c8烃滴加到溶液之前或过程中加入晶种。晶种可以通过将式ⅰ化合物溶于正丙醇,滴加正庚烷,在低温下析晶过滤得到。其产物可用过滤、离心等方法分离,分离后的产物可用空气干燥、鼓风干燥、真空干燥等方法干燥。
[0022]
与现有技术相比,本发明的优点在于:本发明采用了一种工艺简单、可重复性高、纯度高且稳定性好、易于保存的中间体(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇晶型a作为原料,制备达格列净,避免了大量杂质的产生,有利于提高产物纯度及收率。
附图说明
[0023]
图1为本发明背景技术中路线1的合成图;
[0024]
图2为本发明背景技术中路线2的合成图;
[0025]
图3为本发明背景技术中路线3的合成图;
[0026]
图4为本发明背景技术中路线4的合成图;
[0027]
图5为本发明背景技术中路线5的合成图;
[0028]
图6为本发明背景技术中路线6的合成图;
[0029]
图7为本发明背景技术中路线7的合成图;
[0030]
图8为本发明背景技术中路线8的合成图;
[0031]
图9为本发明实施例1中式ⅰ化合物的hplc图谱;
[0032]
图10为本发明实施例1中化合物ⅰ的x射线衍射图谱;
[0033]
图11为本发明实施例1中化合物ⅰ的红外光谱检测图谱;
[0034]
图12为本发明实施例1中化合物ⅰ的紫外光谱检测图谱;
[0035]
图13为本发明实施例1中化合物ⅰ的核磁共振氢谱检测图谱;
[0036]
图14为本发明实施例1中化合物ⅰ的核磁共振碳谱检测图谱。
具体实施方式
[0037]
以下结合附图实施例对本发明作进一步详细描述。
[0038]
实施例1:
[0039]
本发明实施例中达格列净的制备方法包括以下步骤:
[0040]
(1)制备式ⅰ化合物(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧苄基)苯基)-6-(羟甲基)-2-丙氧四氢-2h-吡喃-3,4,5-三醇
[0041]
称取10.0g(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧基苄基)苯基)-6-(羟甲基)四氢-2h-吡喃-2,3,4,5-四醇(hplc纯度83.46%)加入反应器中,室温下加入50ml正丙醇将其搅拌溶清,加入1.0g甲磺酸,室温搅拌10小时后,加入饱和碳酸钠溶液淬灭反应;然后加入乙酸乙酯分层萃取,有机层水洗后减压浓缩得到式ⅰ化合物油状物,(hplc纯度79.33%,ms为407)。
[0042]
(2)式ⅰ化合物晶型a的制备
[0043]
称取10.0g(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧基苄基)苯基)-6-(羟甲基)-2-丙氧基四氢-2h-吡喃-3,4,5-三醇(hplc纯度79.33%)加入反应器中,室温下加入20ml正丙醇将样品搅拌溶清,加入3.0g水,滴加200ml正庚烷后,加入晶种,室温搅拌10小时后,将悬浊液过滤、干燥,即可得到固体晶型a,如图9~14所示,hplc纯度99.37%。
[0044]
(3)达格列净的制备
[0045]
称取10.0g晶型a(hplc纯度99.37%)加入反应器中,加入30ml二氯甲烷和30ml乙腈,加入14g三乙基硅烷,降温至-20℃,然后滴加三氟化硼乙醚溶液12g,滴加完毕后保温10小时,加入饱和碳酸钠溶液淬灭反应,水层经过二氯甲烷萃取后与有机层合并,然后水洗两次,经无水硫酸钠干燥后减压浓缩得到约7.8g类白色固体(hplc纯度99.07%)。
[0046]
实施例2:
[0047]
本实施例提供一种式ⅰ化合物晶型a的制备方法,包括以下步骤:
[0048]
称取10.0g式ⅰ化合物(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧基苄基)苯基)-6-(羟甲基)-2-丙氧基四氢-2h-吡喃-3,4,5-三醇(纯度为纯度79.33%)加入反应器中,室温下加入30ml正丙醇将样品搅拌溶清,滴加400ml正庚烷后,加入晶种,室温搅拌10小时后,将悬浊液过滤、干燥,即可得到目标产物,如图2、3、4、5、6所示;如图1所示,纯度为99.22%。
[0049]
实施例3:
[0050]
本实施例提供一种式ⅰ化合物晶型a的制备方法,包括以下步骤:
[0051]
称取10.0g(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧基苄基)苯基)-6-(羟甲基)-2-丙氧基四氢-2h-吡喃-3,4,5-三醇(hplc纯度79.33%)加入反应器中,室温下加入10ml正丙醇,升至50℃将样品搅拌溶清,等降至0℃后,加入3.0g水,滴加200ml正庚烷后,加入晶种,室温搅拌10小时后,将悬浊液过滤、干燥,即可得到固体晶型a(hplc99.26%)。
[0052]
实施例4:
[0053]
本实施例提供一种式ⅰ化合物晶型a的制备方法,包括以下步骤:
[0054]
称取10.0g(2s,3r,4s,5s,6r)-2-(4-氯-3-(4-乙氧基苄基)苯基)-6-(羟甲基)-2-丙氧基四氢-2h-吡喃-3,4,5-三醇(hplc纯度79.33%)加入反应器中,室温下加入20ml正丙醇,升至50℃将样品溶清,等降至室温后加入2.0g水,滴加100ml正庚烷后,加入晶种,然后再滴加100ml正庚烷,室温搅拌10小时后,将悬浊液过滤、干燥,即可得到固体晶型a(hplc99.41%)。