乙氧基化甘油酯及其制备方法
1.本发明涉及特定类型的钙催化剂用于制备烷氧基化甘油酯的用途,在这样的催化剂存在下制备的烷氧基化甘油酯和制备烷氧基化甘油酯的方法。
2.在文献中已知烷氧基化的脂肪酸酯在不同领域例如家庭护理、化妆品、纺织和其它工业中作为表面活性剂。经常,这样的烷氧基化酯的制备在催化剂的存在下进行。通常采用的催化剂是例如氢氧化钠/醇混合物或醇钠。
3.de-a 3 914 131公开了在烷氧基化脂肪酸酯的制备中使用镁/铝盐作为催化剂。在us 4,820,673中描述了钙基催化剂用于有机化合物例如脂肪醇的烷氧基化。us 4,835,321提供了钙/铝基催化剂用于烷氧基化脂肪醇的合成。
4.us 5,386,045公开了使用由烷氧基化的醇、含钙化合物、路易斯酸性金属和无机酸化合物的金属醇盐制备的催化剂,或由含钙化合物和特定活化剂制备的催化剂,用于制备单醇、二醇或三醇的烷基化脂肪酸酯。
5.本发明的一个目标是提供具有特别低的分解程度的乙氧基化甘油酯,其特征在于少量的游离羟基。
6.现已令人惊讶地发现了特定类型的钙催化剂特别可用于实现这一目标。
7.因此,本发明提供可通过包括以下的反应获得的催化剂(c)的用途:
8.(a)氢氧化钙,和
9.(b)包含3至40个碳原子的羧酸,
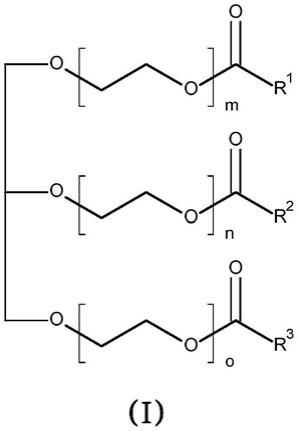
10.用于由环氧乙烷和一种或多种甘油三酯制备通式(i)的乙氧基化甘油酯,
[0011][0012]
其中r1、r2和r3相同或不同,并独立地选自饱和或不饱和的线性或支化的具有3至40个碳原子的烷基链;和
[0013]
m、n和o相同或不同,并且每个独立地为整数,前提是m+n+o之和的数量平均为至少3,
[0014]
其中在催化剂(c)的制备中氢氧化钙(a)和羧酸(b)的摩尔比为1:1-1:5。
[0015]
本发明还提供通式(i)的乙氧基化甘油酯
[0016][0017]
其在如以上限定的钙催化剂(c)存在下,由环氧乙烷和一种或多种式(ii)的甘油三酯制备
[0018][0019]
特征在于
[0020]
式(i)和(ii)中的r1、r2和r3相同或不同,并独立地选自饱和或不饱和的线性或支化的c
7-c
24
烷基链;式(i)中的m、n和o相同或不同,并且每个独立地为1-200的整数,前提是m+n+o之和的数量平均大于5。
[0021]
虽然式中m、n和o是整数,但是描述的产物通常是各种组分的混合物。
[0022]
本发明还提供制备式(i)的乙氧基化甘油酯的方法,
[0023][0024]
包括以下步骤
[0025]
i)引入如以上限定的催化剂(c)和式(ii)的甘油三酯
[0026][0027]
至耐压反应器中;
[0028]
ii)任选地用氮气或其它保护气体代替反应器中的空气;
[0029]
iii)任选地在50-200℃的温度和/或小于0.8巴的压力下干燥反应器内容物;
[0030]
iv)加热反应器的内容物至80℃-200℃的温度;
[0031]
v)任选地用氮气或其它保护气体将反应器加压至大于大气压力0.3巴-3.5巴的压力。
[0032]
vi)用环氧乙烷气体加压反应器至大于大气压力1.5巴-10巴的压力,前提是压力大于步骤vi)前的压力;
[0033]
vii)使混合物反应直至反应器中的压力恒定;
[0034]
其中式(i)和(ii)中的r1、r2和r3相同或不同,并独立地选自饱和或不饱和的线性或支化的c
7-c
24
烷基链;
[0035]
式(i)中的m、n和o相同或不同,并且每个独立地为1-200的整数,前提是m+n+o之和的数量平均大于5。
[0036]
本发明的另一方面是如以上描述的乙氧基化甘油酯在餐具清洁组合物中的用途。
是饱和或不饱和的线性或支化的c
8-c
18
烷基基团,r5、r6、r7和r8是氢和p是2-5的整数。
[0050]
可取的是在具有pka值为3或更小、优选2或更小、优选0或更小,和经常-3或更小的酸(ac)的存在下进行获得催化剂(c)的反应。
[0051]
优选地,酸(ac)选自硫氧化物和磷氧化物的酸,更优选选自硫酸、亚硫酸、磺酸(例如甲磺酸)、磷酸(phosphorus acid)、亚磷酸和膦酸(例如甲膦酸)。硫酸、亚硫酸和甲磺酸特别令人感兴趣。
[0052]
在特别优选的实施方案中,在硫酸的存在下进行获得催化剂(c)的反应。
[0053]
优选地,在反应中使用酸(ac),使得氢氧化钙(a)与酸(ac)的摩尔比为5:1-1:1、更优选3:1-1:1、甚至更优选2:1-1:1。
[0054]
特别有利的是如下制备钙催化剂(c):首先使氢氧化钙(a)与羧酸(b)反应,优选在如以上描述的溶剂中反应,这之后用酸(ac)进一步处理反应混合物。
[0055]
对于获得钙催化剂(c)的反应而言,可以采用任何常见的反应器,优选具有搅拌/混合装置例如磁力搅拌器、机械搅拌器、静态混合器、共混器或间歇式分散器的反应器。优选地,使用间歇式分散器进行组分的混合。
[0056]
优选在0.5-2巴、更优选0.8-1.5巴、甚至更优选0.9-1.2巴的压力下进行催化剂(c)的制备。在优选实施方案中,在大气压力下制备催化剂。此外,优选在从-30℃至80℃、优选从-10℃至60℃、更优选从0℃至50℃的温度下制备催化剂(c)。在优选的实施方案中,在20-40℃的温度下,尤其是在室温下制备催化剂。
[0057]
如此制备的钙催化剂(c)通常具有在0.5重量%和5重量%之间、经常1-4重量%、经常2-3重量%的ca
2+
离子含量。任选地,催化剂可以通过采用常用的方法脱去易挥发组分,例如溶剂、水和其它挥发副产物。优选地,在真空中例如在小于0.8巴、优选小于0.3巴、更优选小于0.1巴的压力下和/或在升高的温度例如50至180℃、优选70至150℃、更优选80至120℃下去除挥发性组分。
[0058]
在特别优选的实施方案中,在旋转蒸发器上在小于0.1巴的压力和80℃-120℃的温度下去除挥发性组分。
[0059]
本发明的乙氧基化甘油酯特别是式(i)的乙氧基化甘油酯
[0060][0061]
并在如以上限定的钙催化剂(c)的存在下由环氧乙烷和一种或多种式(ii)的甘油三酯制备
[0062][0063][0064]
本发明的乙氧基化甘油酯的式(i)和制备本发明的乙氧基化甘油酯的一种或多种式(ii)的甘油三酯中的r1、r2和r3相同或不同,并独立地选自饱和或不饱和的线性或支化的c
7-c
24
烷基链,优选c
9-c
20
烷基链,更优选c
12-c
18
烷基链。
[0065]
本发明的乙氧基化甘油酯的式(i)中的m、n和o每个独立地为1-200、优选1至80、更优选2至70的整数,前提是m+n+o之和的数量平均大于5、优选大于8、更优选8至200、甚至更优选8至80,由根据din en iso 3681测量的皂化值计算。
[0066]
一种或多种式(ii)的甘油三酯没有特别限制,并可以是天然的甘油三酯或合成的甘油三酯。优选地,甘油三酯不含有任何游离的羟基基团。
[0067]
在以上限定的催化剂(c)的存在下制备的本发明的乙氧基化甘油酯的羟基值(根据din en iso 4629-2测量)通常比一种或多种式(ii)的甘油三酯的羟基值高不到6mg koh/g。
[0068]
经常,本发明的乙氧基化甘油酯的羟基值总计小于7mg koh/g、优选小于6mg koh/g、更优选小于5mg koh/g。此外,本发明的乙氧基化甘油酯中ch2oh基团与烷基-ch3基团之比通常小于0.15、优选小于0.12、更优选小于0.08、甚至更优选小于0.06,以质子-nmr谱中相应信号的积分之比测量。在特别优选的实施方案中,本发明的乙氧基化甘油酯的羟基值小于7mg koh/g,并且本发明的乙氧基化甘油酯的ch2oh基团与烷基-ch3基团之比小于0.12。在更特别优选的实施方案中,羟基值小于5mg koh/g,并且ch2oh基团与烷基-ch3基团之比小于0.06。
[0069]
优选地,本发明的乙氧基化甘油酯的皂化值小于220mg koh/g、更优选小于150mg koh/g。
[0070]
制备如以上描述的乙氧基化甘油酯的本发明方法包括以下步骤
[0071]
i)引入如以上限定的催化剂(c)和如以上描述的一种或多种式(ii)的甘油三酯至耐压反应器中;
[0072]
ii)任选地用氮气或其它保护气体代替反应器中的空气;
[0073]
iii)任选地在50-180℃的温度和/或小于0.8巴的压力下干燥反应器内容物;
[0074]
iv)加热反应器的内容物至80℃-200℃的温度;
[0075]
v)任选地用氮气或其它保护气体将反应器加压至1.3-3.5巴的压力。
[0076]
vi)用环氧乙烷加压反应器至1.5巴-10巴的压力,前提是压力大于步骤vi)前的压力;和
[0077]
vii)使混合物反应直至反应器中的压力恒定。
[0078]
在步骤i)中,催化剂(c)可以从以上描述的制备反应中直接获得的形式引入,或以脱去了挥发性化合物的形式引入。式(ii)的甘油三酯可以以它们原始形式引入或者可以在使用前纯化。优选地,在使用前纯化式(ii)的甘油三酯,由此从起始材料分离痕量的甘油二酯、甘油单酯、游离的甘油和其它杂质。
[0079]
优选将钙催化剂(c)以0.1-5重量%、优选0.2-3重量%、更优选0.3-2重量%的量引入反应器,基于式(ii)的甘油三酯和环氧乙烷的混合物的总重量。
[0080]
耐压反应器没有特别限制但被设计为耐受方法中采用的压力,因此在方法期间不会被损坏。优选地,反应器被设计为耐受大于10巴、更优选大于15巴且小于0.01巴、更优选小于0.001巴的压力。优选地,耐压反应器是高压釜,更优选配备有搅拌装置例如磁力或机械搅拌器的高压釜。
[0081]
通常,不一定需要用氮气或其它保护气体代替反应器中的空气,因为本发明的乙氧基化甘油酯将至少部分在该方法中产生。然而,反应器中的空气,特别是氧气可以导致通常在烷氧基化反应期间的安全问题和由于所用材料和产生的产物的氧化和/或水解所致的分解产物,尤其是在升高的温度下。因此,可取的是在步骤i)之后进行本发明方法的步骤ii)。
[0082]
通常,也不一定需要干燥反应器内容物的步骤,因为本发明的乙氧基化甘油酯将至少部分在该方法中产生。然而,水和醇可以促进所用材料和在反应条件下产生的产物的水解和酯交换。尤其是如果在步骤i)中将钙催化剂(c)以从以上描述的制备反应中直接获得的形式引入反应器,可取的是进行干燥步骤,因为直接获得的催化剂(c)通常含有极性溶剂的残留物或它们与水的混合物。在钙催化剂(c)在将被引入反应器之前脱去挥发性组分的情况下,可以省略干燥步骤iii)。然而,在这种情况下可取的是进行步骤iii),因为挥发性组分也可以作为一种或多种式(ii)的甘油三酯中的杂质存在。因此,在特别优选的实施方案中,进行步骤iii)。
[0083]
干燥反应器内容物的步骤iii)通常在50℃-180℃、优选60℃-150℃、优选70℃-130℃、经常80℃-120℃的温度下,和在小于0.8巴、优选小于0.1巴、更优选小于0.05巴的压力下进行。如此产生的真空优选是动态真空。
[0084]
用于产生真空的真空泵没有特别限制;然而优选使用用于产生真空的抽气机。此外,可取的是逐渐提高反应器中温度和减小压力以防止沸腾延迟。在特别优选的实施方案中,干燥反应器内容物的步骤在80℃-120℃的温度和小于0.01巴的压力下,优选在至少15分钟的时间段内、更优选在至少30分钟的时间段内、甚至更优选在至少1小时的时间段内进行。特别优选干燥反应器的内容物至恒定质量。
[0085]
在干燥步骤iii)之后,中断在真空泵和反应器之间的流体管线,以确保添加至反应器的组分在干燥之后保持在反应器中并且没有从其中直接抽出。此外,优选在进行另外的步骤之前用氮气或其它的保护气体补偿反应器中的真空,以减小空气进入反应器的风险。
[0086]
加热反应器的内容物的步骤iv)通常在80℃-200℃、优选120℃-190℃、更优选160℃-180℃的温度下进行。维持这个温度至少直至步骤vi)完成,优选直至步骤vii)完成。
[0087]
在设置步骤iv)中的温度之后,可以任选地在步骤v)中将反应器用氮气或其它保
护气体加压至大于大气压力0.3-3.5巴、优选0.5-2.0巴、优选0.7-1.5巴、更优选0.8-1巴的压力。通过进行这个步骤v),用保护气体稀释在以下步骤中引入的环氧乙烷,由此促进环氧乙烷进入反应器的压力控制剂量。
[0088]
在步骤vi)中,将反应器进一步用环氧乙烷加压至总内部压力为1.5-10巴、优选2-8巴、更优选3-6巴、更优选4-5巴,前提是步骤vi)中的压力大于步骤vi)之前的压力。
[0089]
在步骤vii)期间,在引入预期量的环氧乙烷之后,关闭环氧乙烷并且使反应进行直至反应器中的压力恒定。
[0090]
在本发明的意义上,如果在15min、优选30min、更优选1小时的时间段内压力改变不大于0.05巴,则认为压力是恒定的。特别优选的是反应器中的压力在1小时的时间段内改变不大于0.01巴。
[0091]
通常,将全部量的环氧乙烷添加至反应器并且在小于1000分钟内,经常小于800分钟内由本发明的方法获得恒定压力。在特别优选的实施方案中,在小于700分钟内获得恒定的压力。这时候,认为环氧乙烷和一种或多种式(ii)的甘油三酯之间的反应完成。
[0092]
在步骤vii)完成之后,可取的是在分离本发明的乙氧基化甘油酯之前从反应器中去除残余的环氧乙烷,以便防止在产物分离之后发生与环氧乙烷的任何不想要的反应。优选地,通过冷却反应器内容物至50-120℃、更优选70-100℃、更优选85-95℃的温度,并且采用小于0.8巴、优选小于0.1巴、更优选小于0.05巴的压力从反应器中去除残余的环氧乙烷。如此产生的真空优选是动态真空。用于产生真空的真空泵没有特别限制;然而优选使用用于产生真空的抽气机。在这些条件下去除残余的环氧乙烷优选进行至少10分钟、优选至少30分钟、更优选至少1小时。
[0093]
分离本发明的乙氧基化甘油酯的方法没有特别限制。然而,优选在升高的温度下,尤其是在50-120℃、优选60-100℃、更优选70-90℃的温度下分离产物。在这些温度下本发明的乙氧基化甘油酯通常为液体状态,并具有足够低的粘度,并因此可以比固态下更容易地从反应器中转移出来,例如通过将产物从反应器中倒出或经由底部阀倒出,由此使反应器中的残余物的量最小。因此,也促进反应器的随后清洁和维护。
[0094]
令人惊讶地发现了使用以上描述的钙催化剂(c)制备根据本发明的乙氧基化甘油酯的方法可以在任何阶段中断,并在之后的时间点继续,而没有明显提高反应时间。与此相反,使用本领域已知的催化剂的制备方法的中断需要在中断之后实质上更长的再活化,从而总反应时间提高较大量的时间。
[0095]
此外,观察到采用本领域已知的催化剂的制备方法导致在反应器中灰白色的固体残余物,如果使用特定类型的钙催化剂(c),没有发现该残余物。以下实施例和权利要求书进一步更详细地阐述本发明。
实施例
[0096]
对比合成例1制备us 5,386,045的钙催化剂
[0097]
在室温下在氮气氛下在高压釜中搅拌125g的醇乙氧基化物(来自c
10
/c
12-脂肪醇和40重量%环氧乙烷,例如来自vista chemical company的alfonic 1012-40)、2g的2-乙基己酸和10.9g的氢氧化钙的混合物,同时在10min的时间段内添加2g的浓硫酸。在完全添加硫酸之后,继续搅拌5h。随后,将混合物加热至150℃并在15min内在氮气流中去除挥发性
组分。将混合物冷却至125℃并添加17.5g的三烷氧基铝(含有约6重量%al并具有10个碳原子的平均烷氧基碳链长度)。
[0098]
将混合物在125℃下搅拌另外的2h,这之后温度升高至190℃并在氮气流中去除挥发性组分。在190℃下另外的0.5小时之后,将混合物冷却至环境温度,提供ca
2+
含量为大约3重量%和al
3+
含量为大约0.6重量%的催化剂(在此之后称为“(c-0)”)。
[0099]
对比合成例2制备助催化剂(单油酸甘油酯)
[0100]
将9.2g的甘油和28.2g的油酸的混合物加热至175℃并在这个温度下搅拌,同时用dean-stark装置去除水,直至酸值《2mg koh/g。
[0101]
合成实施例1
[0102]
用式(iii)的羧酸制备钙催化剂(c)
[0103]
a)在环境温度下用间歇式分散器(来自ika werke gmbh&co kg的ultra turrax)搅拌1047.0g的式(iii)的羧酸(由clariant produkte(德国)gmbh以商品名“emulsogen col 050”销售)、55.8g的氢氧化钙和360.6g的丙-2-醇的混合物5min。在这之后,在两分钟内添加44.2g的浓硫酸并用间歇式分散器再次搅拌混合物5min,提供ca
2+
含量为2.00重量%的催化剂(在此之后称为“(c-1)”)。
[0104]
可通过使用甲磺酸或亚硫酸代替硫酸获得提供ca
2+
含量为大约2.00重量%的催化剂的类似结果。
[0105]
b)在环境温度下用间歇式分散器(来自ika werke gmbh&co kg的ultra turrax)搅拌1047.0g的式(iii)的羧酸(由clariant produkte(德国)gmbh以商品名“emulsogen col 050”销售)、55.8g的氢氧化钙和360.6g的丙-2-醇的混合物5min。在这之后,在两分钟内添加42.9g的甲磺酸(99重量%)并用间歇式分散器再次搅拌混合物5min,提供ca
2+
含量为2.00重量%的催化剂(在此之后称为“(c-3)”)。
[0106]
c)在环境温度下用间歇式分散器(来自ika werke gmbh&co kg的ultra turrax)搅拌1047.0g的式(iii)的羧酸(由clariant produkte(德国)gmbh以商品名“emulsogen col 050”销售)、55.8g的氢氧化钙和360.6g的丙-2-醇的混合物5min。在这之后,在两分钟内添加603.7g的亚硫酸(6重量%)并用间歇式分散器再次搅拌混合物5min。在真空下去除溶剂混合物,提供ca
2+
含量为大约2重量%的催化剂(在此之后称为“(c-4)”)。
[0107]
emulsogen col 050是商购产品羧酸(b),包含由式(iii)表示的羧酸作为主要组分,其中r4是油烯基,r5、r6、r7和r8是氢,和p为5。
[0108]
合成实施例2
[0109]
用式(iv)的羧酸制备钙催化剂(c)
[0110]
用间歇式分散器(来自ika werke gmbh&co kg的ultra turrax)搅拌114g异壬酸、26.7g氢氧化钙、346.38g的丙-2-醇和26.7g水的混合物5min。在这之后,一次添加10.62g的浓硫酸并用间歇式分散器再次搅拌混合物5min,提供ca
2+
含量为2.75重量%的催化剂(在此之后称为“(c-2)”)。
[0111]
合成实施例3
[0112]
一般烷氧基化程序
[0113]
将式(ii)的甘油三酯、催化剂和如果适用的助催化剂置于玻璃高压釜中,然后通过交替地施加真空和引入氮气(3个循环)将其用氮气冲洗。在抽气机真空下在100℃下干燥
混合物1小时。用氮气将高压釜中的压力恢复至环境并加热至175℃。在这个温度下将高压釜用氮气加压至大于大气压力0.8巴的压力,这之后进行环氧乙烷的压力控制剂量直至大于大气压力4.5巴的最大压力。
[0114]
以半间歇方法使用环氧乙烷的自动化剂量在给定温度窗口内进行乙氧基化直至规定的最大压力。根据容器的提高的填充体积调节压力。在引入预期量的环氧乙烷并关闭环氧乙烷入口之后,继续反应直至压力变得恒定。
[0115]
将反应器内容物冷却至90℃并且施加抽气机真空30min以便去除残余的环氧乙烷。将温度降低至80℃并将最终产物转移至储存容器中并分析。典型的批料规模是400g至2000g。通过重力法并通过根据din en iso 3681测定皂化值来确定预期量的环氧乙烷的摄取。
[0116]
在以下表1(摩尔当量)中显示合成实施例3中采用的材料和到恒定压力的反应时间:
[0117]
[0118][0119]
使用c-0时中断合成实施例3持续15小时导致反应以比开始时反应的引发明显更慢的速率重新引发。使用c-1或c-2时中断导致反应以与在中断前直接观察的基本上相同的
速率重新开始。
[0120]
此外,乙氧基化还可通过使用相同的程序用催化剂c-1或c-2使用脂肪醇、脂肪酸烷基酯和脂肪酸亚烷基二醇二酯代替甘油三酯来进行。此外,还可以在另外的甘油的存在下进行甘油三酯、脂肪醇、脂肪酸烷基酯和脂肪酸亚烷基二醇二酯的乙氧基化,以获得由于产物中较大量的羟基基团所致具有较高极性的产物。
[0121]
从表1显然可见,在本发明实施例中,采用催化剂(c-1)或(c-2)时,甘油三酯以比在采用催化剂(c-0)或kotbu(任选地使用单油酸甘油酯作为助催化剂)的对比例中明显更高的速率反应。在引入任何量的环氧乙烷和采用任何甘油三酯的情况下,观察到更快的反应速率。
[0122]
此外,在采用催化剂(c-0)的反应之后清空高压釜之后,灰白色固体附着至搅拌器和温度传感器以及在反应容器中。当在反应中采用催化剂(c-1)或催化剂(c-2)时没有观察到这些固体。
[0123]
在表2中,描述在不同条件下几个对比例和本发明实施例的外观。
[0124]
表2:
[0125][0126]
[0127]
在表3中,显示几个本发明实施例和对比例的羟基值和ch2oh基团与烷基-ch3基团之比。
[0128]
根据din en iso 4629-2测量羟基值。由使用bruker nmr分光光度计采用400mhz和cdcl3作为溶剂的相应的质子-nmr信号的积分比计算ch2oh基团与烷基-ch3基团之比。
[0129]
来自采用(c-0)作为催化剂的对比例的灰白色固体残余物是不溶的并且没有分析。
[0130]
表3:
[0131][0132][0133]
从对比例和本发明实施例的oh值显然可见,本发明实施例使用催化剂(c-1)的乙氧基化甘油三酯在合成反应期间比使用催化剂(c-0)或kotbu的对比例的乙氧基化甘油三酯经历更少的分解。
[0134]
特别地,在乙氧基化之后酯基的皂化显示以较低的程度发生,这从衍生自聚乙氧基-oh的ch2oh基团与衍生自脂肪酸烷基的烷基-ch3基团之比看出,所述聚乙氧基-oh产生自不希望的副反应(例如由于较长的反应时间和高反应温度)。
[0135]
根据实施例2、对比3和对比4的乙氧基化甘油三酯(各由1摩尔当量的椰子油和
22.5摩尔当量的环氧乙烷制备)在水中的溶解度通过在25℃下在玻璃试管中混合0.5重量份的相应乙氧基化甘油三酯与99.5重量份的去离子水来检测,并在混合后立即和混合后1小时对所得组合物的透明度进行视觉评价。在每种情况下,1小时时间段内改变不明显。根据以下评级进行视觉评价:
[0136]
清澈
ꢀꢀꢀꢀꢀ
没有观察到悬浮物质
[0137]
几乎清澈 观察到痕量悬浮物质
[0138]
稍微浑浊 观察到悬浮物质,深色背景依然可见
[0139]
浑浊
ꢀꢀꢀꢀꢀ
悬浮物质明显,深色背景几乎不可识别
[0140]
不透明
ꢀꢀꢀ
悬浮物质非常明显,深色背景不可识别。
[0141]
评价示于表4中:
[0142]
表4:
[0143]
样品获自混合后立即混合后1小时实施例2稍微浑浊稍微浑浊对比3不透明不透明对比4几乎清澈几乎清澈
[0144]
从以上结果可看出,使用催化剂c-1制备的本发明乙氧基化甘油酯(实施例1)具有比使用本领域已知的催化剂c-0制备的对比产物(对比3)明显更好的水中溶解度。这对于例如产物作为表面活性剂的适用性是重要的。
[0145]
尽管游离羟基的量较小,但观察到改善的溶解度,这预期由于较高的极性所致促进在水中溶解。
[0146]
上述区别与采用催化剂c-0的乙氧基化过程中不溶性固体残余物的形成一致。
[0147]
由kotbu和助催化剂制备的产物(对比4)的溶解度提高是大量携带oh基的分解产物的结果,这提高了混合物的极性并因此提高了组合物在水中的总溶解度。
[0148]
应用实施例1:干燥能力和清洁餐具清洁机内部
[0149]
调查了包含实施例4的乙氧基化甘油酯的机洗餐具洗涤剂组合物f2的干燥能力。作为对比例,测试包含改性脂肪醇乙氧基化物的对比制剂f1的干燥能力。
[0150]
测试条件:
[0151]
餐具清洁机:miele g 1222 sc gsl-2
[0152]
测试用具餐具:10个头盘匙
[0153]
10个头盘叉
[0154]
10个茶匙
[0155]
2个布菜匙
[0156]
12个饮用玻璃杯
[0157]
10个瓷杯
[0158]
25个瓷盘
[0159]
3个san(聚苯乙烯-共聚-丙烯腈)盘
[0160]
3个pp(聚丙烯)盘
[0161]
6个pp碗
[0162]
餐具清洁程序:p4r0无预漂洗
[0163]
50℃下主要漂洗
[0164]
65℃下最后漂洗
[0165]
水硬度:
ꢀꢀꢀꢀꢀ
21
°
dh
[0166]
水软化:
ꢀꢀꢀꢀꢀ
无
[0167]
清洁剂剂量: 18g,在打开量斗后立即添加至清洁剂托盘中
[0168]
污染物:
ꢀꢀꢀꢀꢀ
50g冰冻污物,在打开量斗后立即添加
[0169]
漂洗助剂:
ꢀꢀꢀ
无
[0170]
清洗周期:
ꢀꢀꢀ4[0171]
所有的物品用软化水、neodisher a 8、柠檬酸和软化水处理一次。
[0172]
评价:
[0173]
在餐具清洁周期完成之后30分钟开始测试用具的评价。在这个时间期间,餐具清洁机门是关闭的。对于每次测试,评价餐具清洁周期2至4。在每种情况下用1000-1500勒克斯的光照进行评价。
[0174]
以固定的顺序和设定的时限,对每个测试用具物品计数残留水的粘附滴数。取决于计数的滴数,对于每个测试用具物品产生以下干燥能力评级结果:
[0175]
瓷器、不锈钢和玻璃的评级:
[0176]
0干燥,没有水滴
[0177]
1 1个水滴
[0178]
2 2个水滴
[0179]
3 3个水滴
[0180]
4 4个水滴
[0181]
5 5个水滴
[0182]
6多于5个水滴
[0183]
塑料件的评级:
[0184]
0干燥,没有水滴
[0185]
1 1个水滴
[0186]
2 2个水滴
[0187]
3 3个水滴
[0188]
4 4个水滴
[0189]
5 5个水滴
[0190]
6 6个水滴
[0191]
7 7个水滴
[0192]
8多于7个水滴。
[0193]
在这个评级方案中,对于每个测试用具餐具,最佳性能分数为0,最差性能分数为6。对于每个餐具清洁周期2、3和4,形成所有测试餐具的分数总和。为了比较制剂f1和f2,在每个情况下将餐具清洁周期2至4的所有总和的平均评级进行平均。这导致最差干燥性能的理论最大值为630并且最佳干燥性能的理论最小值为0。
[0194]
结果显示在以下表5中。
[0195]
此外,评价餐具清洁机的塑料零件(过滤器、漂洗助剂腔)上的脂肪残余(以1至7的
评级,其中1表示大量的残余物和7表示没有残余物)。这些结果也显示在表5中。
[0196]
组成:
[0197]
在以下表5中显示制剂f1和f2的组成。
[0198]
应用实施例2:机洗餐具洗涤剂组合物的漂洗辅助性能
[0199]
调查了根据本发明的制剂f2的漂洗辅助性能。作为对比例,测试了对比制剂f1的漂洗辅助性能。
[0200]
测试条件:
[0201]
餐具清洁机:
ꢀꢀꢀꢀꢀꢀ
miele g 1222sc gsl
[0202]
测试用具餐具:
ꢀꢀꢀꢀ
6个饮用玻璃杯(较高品质)
[0203]
(8个材料组)
ꢀꢀꢀꢀꢀꢀ
6个饮用玻璃杯(较低品质)
[0204]
3个pp碗
[0205]
3个密胺盘
[0206]
3个黄油碟+4把刀(不锈钢;较低品质)
[0207]
4把刀(不锈钢;较高品质)
[0208]
3个瓷盘(较高品质)
[0209]
3个瓷盘(较低品质)
[0210]
餐具清洁程序:程序4,r=2无预漂洗
[0211]
50℃下主要漂洗
[0212]
65℃下最后漂洗
[0213]
水硬度:
ꢀꢀꢀꢀꢀ
21
°
dh
[0214]
水软化:
ꢀꢀꢀꢀꢀ
无
[0215]
清洁剂剂量: 18g,在开始测试前添加至量斗
[0216]
污染物:
ꢀꢀꢀꢀꢀ
100g冰冻污物,在打开量斗后立即添加
[0217]
漂洗助剂:
ꢀꢀꢀ
无
[0218]
清洗周期:
ꢀꢀꢀ4[0219]
所有测试用具餐具(除了pp碗之外)用软化水、neodisher a 8、柠檬酸和再次用软化水处理一次。
[0220]
评价:
[0221]
在餐具清洁周期完成后打开餐具清洁机门之后至少60分钟开始测试用具的评价。对于每次测试,评价餐具清洁周期2至4。根据以下评级进行评价:
[0222]
考虑视觉评级的漂洗助剂效果:
[0223]
污渍
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
具有不同尺寸和强度的污渍
[0224]
接触斑点
ꢀꢀꢀꢀꢀ
由测试用具餐具和餐具清洁机的零件之间的接触点产生的污渍
[0225]
条纹
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
漂洗助剂条纹
[0226]
成膜
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
在测试用具餐具上均匀铺开的连续膜
[0227]
结构化膜形成 分散裂开的膜
[0228]
固体残余
ꢀꢀꢀꢀꢀ
固体粉末或结晶残余物
[0229]
脂肪残余
ꢀꢀꢀꢀꢀ
脂肪滴或脂肪膜形成
[0230]
虹彩
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
闪烁的虹彩
[0231]
视觉评级分数:
[0232]
10完美
[0233]
9 完美至几乎看不见
[0234]
8 几乎看不见
[0235]
7 几乎看不见至可见
[0236]
6 可见
[0237]
5 可见至妨碍的
[0238]
4 妨碍的
[0239]
3 妨碍的至不可接受
[0240]
2 不可接受
[0241]
1 绝对不可接受
[0242]
根据以上视觉评级分数,以上列出的八种漂洗助剂效果的组合导致1-10的评级,其中1的评级表示最差的性能和10的评级表示最好的性能。对于每个餐具清洁周期中的以上8个测试用具材料组中的每个,确定平均评级,之后计算每个单独餐具清洁周期中的所有材料组的评级之和,之后确定餐具清洁周期2至4整体的平均评级。所得平均评级用作制剂f1和f2的最终漂洗辅助性能。这导致最好性能的理论最大值为80和最差性能的理论最小值为8。
[0243]
组成:
[0244]
在以下表5中显示根据本发明的制剂f2的组成和f1的组成。结果也显示在表5中。
[0245]
表5:制剂f1和f2的组成、干燥能力、脂肪残余和漂洗辅助性能
[0246]
[0247][0248]
*
)
根据它们的活性组分含量以重量%计添加成分。
[0249]
**
)
硫酸钠作为填料加入,用于使洗涤剂组合物的质量恒定平衡,而不起作用,也不影响洗涤剂组合物的性能。
[0250]
从以上表5的结果明显看出,与组合物f1相比,使用机洗餐具洗涤剂组合物f2导致干燥能力、机器隔室中的脂肪残余和漂洗辅助性能的有益值。
[0251]
此外,制剂f2显示优异的清洁性能和优异的过滤器清洁性质。