1.本发明涉及医药技术领域,具体涉及一种青蒿琥酯衍生物及其制备方法和应用。
背景技术:
2.青蒿琥酯(artesunate,art)是我国自主开发研制的具有倍半萜结构的青蒿素类抗疟特效药,近年研究发现其对白血病、直肠结肠癌、口腔癌、肺癌、胰腺癌、黑色素瘤、乳腺癌、卵巢癌、前列腺癌、中枢神经系统肿瘤及肾癌细胞等都具有抑制作用,美国国家癌症研究所已将其纳入抗癌药物筛选与抗癌活性研究计划中。抗肿瘤机制研究表明青蒿琥酯的过氧桥结构,能够产生氧自由基,提高肿瘤细胞内的活性氧,诱导肿瘤细胞铁死亡和诱导肿瘤细胞凋亡。尽管青蒿琥酯药理作用广泛,具有独特的抗肿瘤机制,但由于水溶性差、半衰期短(~30min)、口服吸收差,导致其生物利用度和抗肿瘤活性比较低。青蒿琥酯对多种肿瘤细胞株的ic50为几十至几百μm,难以单独开发成抗肿瘤药物。因此,寻求生物利用度和抗肿瘤活性高的青蒿琥酯衍生物仍然是人们关注的热点。
技术实现要素:
3.针对目前现有的抗肿瘤药物生物利用度和抗肿瘤活性低的问题,本发明提供一种青蒿琥酯衍生物及其制备方法和应用。
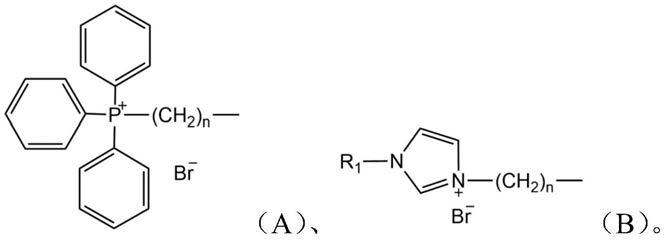
4.为了实现上述目的,本发明第一方面提供一种青蒿琥酯衍生物,该青蒿琥酯衍生物具有式(m)所示的结构,
[0005][0006]
其中,l为式(a)或式(b)所示的基团,且式(a)或式(b)中,n各自独立地为6-20的整数;式(b)中,r1为c1-c6的烷基,
[0007][0008]
本发明第二方面提供一种制备青蒿琥酯衍生物的方法,其特征在于,该方法包括以下步骤:在碱金属盐和第一溶剂的存在下,使式v所示的化合物或式viii所示的化合物与青蒿琥酯接触进行取代反应,
[0009][0010]
其中,且式v或式viii中,n各自独立地为6-20的整数;式viii中,r1为c1-c6的烷基。
[0011]
本发明第三方面提供一种前述青蒿琥酯衍生物在制备抗肿瘤药物中的应用。
[0012]
本发明第四方面提供一种前述青蒿琥酯衍生物或前述方法在抗肿瘤细胞中的应用。
[0013]
与现有技术相比,本发明提供的青蒿琥酯衍生物具有全新的骨架结构和优异的抗肿瘤活性,能够用于制备抗肿瘤药物。该青蒿琥酯衍生物能够充分利用青蒿琥酯独特的抗肿瘤机制和亲脂性阳离子的靶向线粒体作用,发挥优异的抗肿瘤效果。另外,本发明提供的制备青蒿琥酯衍生物的路线简单、原料易得,合成方法容易实现。
具体实施方式
[0014]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0015]
本发明第一方面提供一种青蒿琥酯衍生物,该青蒿琥酯衍生物具有式(m)所示的结构,
[0016][0017]
其中,l为式(a)或式(b)所示的基团,且式(a)或式(b)中,n各自独立地为6-20的整数(如6、7、8、9、10、11、12、13、14、15、16、17、18、19、20);式(b)中,r1为c1-c6的烷基(不限于直链或支链),
[0018][0019]
根据本发明的一些实施方式,该青蒿琥酯衍生物具有式(m’)所示的结构,
[0020][0021]
根据本发明的一些实施方式,式(a)或式(b)中,n各自独立地为6-12的整数。
[0022]
根据本发明的一些实施方式,式(b)中,r1为c1-c3的烷基(如甲基、乙基、正丙基或异丙基)。
[0023]
根据本发明的一些实施方式,所述青蒿琥酯衍生物优选自以下化合物中的一种,
[0024][0025]
优选地,所述青蒿琥酯衍生物选自以下化合物中的一种,
[0026][0027]
本发明第二方面提供一种制备青蒿琥酯衍生物的方法,该方法包括以下步骤:在碱金属盐和第一溶剂的存在下,使式v所示的化合物或式viii所示的化合物与青蒿琥酯(式vi所示的化合物)接触进行取代反应,
[0028][0029]
其中,且式v或式viii中,n各自独立地为6-20的整数;式viii中,r1为c1-c6的烷
基。
[0030]
根据本发明优选的实施方式,式v或式viii中,n各自独立地为6-12的整数(如6、7、8、9、10、11、12)。式viii中,r1为c1-c3的烷基。
[0031]
本发明中,所述取代反应的条件可以包括:温度为60-100℃,优选为70-85℃,时间为4-20h,优选为6-12h。
[0032]
本发明中,所述取代反应中,式v所示的化合物或式viii所示的化合物、青蒿琥酯以及碱金属盐的摩尔比可以为1:(1-2):(1.5-3),优选为1:(1.2-1.6):(2-2.5)。
[0033]
本发明对所述碱金属盐的种类没有特别的限制,只要能够满足本发明的需求即可,优选情况下,所述碱金属盐可以选自碳酸钾(优选指无水碳酸钾)、碳酸钠和碳酸铯中的至少一种。
[0034]
本发明中,相对于1g的式v所示的化合物或式viii所示的化合物,所述第一溶剂的用量可以为30-90ml,优选为40-60ml。
[0035]
本发明对所述第一溶剂的种类没有特别的限制,只要能够满足本发明的需求即可。例如,所述第一溶剂可以选自乙腈、n,n-二甲基甲酰胺、1,4-二氧六环和四氢呋喃中的至少一种。
[0036]
本发明中,所述取代反应在惰性气氛下进行。所述惰性气氛优选由氮气提供。
[0037]
本发明中,对所述取代反应的后处理没有特别的限制,只要能够满足本发明的需求即可,例如可以按照以下步骤进行:将反应结束后的体系进行固液分离(例如过滤),弃去固体,将固液分离得到的液体进行浓缩脱溶,得到脱溶后的剩余物,之后采用溶解溶剂(优选为二氯甲烷)将脱溶后的剩余物溶解之后,再依次经饱和碳酸氢钠溶液、水洗涤(洗涤的次数可以为2-3次),收集有机相,无水硫酸镁干燥;最后经柱层析分离,得到式(m’)所示的目标产物。其中,浓缩的条件没有特别的限制,可以参照本领域常规的方式进行。所述柱层析的过程中,优选采用二氯甲烷和甲醇作为洗脱剂,其中,二氯甲烷和甲醇的体积比可以为(5-25):1,优选为(10-20):1。
[0038]
本发明中,式v所示的化合物或式viii所示的化合物通过自制得到。
[0039]
根据本发明的一些实施方式,所述方法还包括按照以下方式制备式v所示的化合物的步骤:在第二溶剂的存在下,使三苯基膦(式iii所示的化合物)与br(ch2)nbr(式iv所示的化合物)进行反应;其中,n=6-12。
[0040]
根据本发明的一些实施方式,所述方法还包括按照以下方式制备式viii所示的化合物的步骤:在第二溶剂的存在下,式vii所示的化合物与br(ch2)nbr进行反应;其中,n=6-12;
[0041][0042]
本发明中,br(ch2)nbr(式iv所示的化合物)优选自1,6-二溴己烷、1,8-二溴辛烷、1,10-二溴癸烷、1,12-二溴十二烷、1,14-二溴十四烷、1,16-二溴十六烷和1,18-二溴十八烷中的一种。
[0043]
根据本发明的一些实施方式,三苯基膦或式vii所示的化合物与br(ch2)nbr反应的
条件可以包括:温度为70-90℃,时间为5-25h。
[0044]
本发明中,所述第二溶剂选自乙腈、n,n-二甲基甲酰胺、1,4-二氧六环和甲苯中的至少一种。
[0045]
为了获得更好的技术效果,三苯基膦或式vii所示的化合物与br(ch2)nbr反应的加料方式优选按照以下方式进行:在70-90℃下,将三苯基膦或式vii所示的化合物溶于所述第二溶剂中,以10-15wt%/min的速率加入(也即每分钟加入三苯基膦或式vii所示的化合物总量的10-15wt%)含br(ch2)nbr的溶液中。优选地,所述含br(ch2)nbr的溶液由br(ch2)nbr溶于第二溶剂中得到。
[0046]
本发明中,三苯基膦或式vii所示的化合物与br(ch2)nbr的摩尔比可以为1:(4-8)。
[0047]
本发明中,所述第一溶剂优选与所述第二溶剂相同。
[0048]
本发明中,对制备式v所示的化合物和式viii所示的化合物的反应的后处理没有特别的限制,只要能够满足本发明的需求即可,例如可以按照以下步骤进行:将反应结束后的体系进行浓缩脱溶,得到脱溶后的剩余物,之后依次经乙酸乙酯和乙醚洗涤(洗涤的次数可以为2-3次),最后经柱层析分离,得到目标产物。其中,浓缩的条件没有特别的限制,可以参照本领域常规的方式进行。所述柱层析的过程中,优选采用二氯甲烷和甲醇作为洗脱剂,其中,二氯甲烷和甲醇的体积比可以为(5-25):1,优选为(10-20):1。
[0049]
本发明对以上制备过程中涉及的反应条件没有特别的限定,本领域技术人员可以根据有机合成领域内的公知常识以及本发明实施例部分提供的具体实例获得适宜的反应条件。
[0050]
本发明中,式(m)所示的化合物中除式(m’)所示化合物之外的其他立体异构体的制备可以参照上述制备式(m’)所示化合物的制备方法进行制备。
[0051]
本发明第三方面提供一种前述青蒿琥酯衍生物在制备抗肿瘤药物中的应用。
[0052]
本发明第四方面提供一种前述青蒿琥酯衍生物或前述方法在抗肿瘤细胞中的应用。
[0053]
本发明提供的青蒿琥酯衍生物对肿瘤细胞的种类没有特别的限制,均具有一定的效果,但是对某些特定肿瘤细胞的抑制效果尤其显著。所述肿瘤选自肝癌、宫颈癌、卵巢癌、乳腺癌和肺癌中的至少一种,优选自肝癌hepg2、宫颈癌hela、卵巢癌a2780、乳腺癌mcf7和顺铂耐药的肺癌a549/ddp中的至少一种,更优选自宫颈癌hela和/或卵巢癌a2780。其中,所述肺癌优选为顺铂耐药的肺癌。
[0054]
本发明还涉及(在体外)抑制肿瘤细胞生长的方法,该方法包括:将本发明的青蒿琥酯衍生物与肿瘤细胞接触。所述肿瘤细胞选自hepg2、hela、a2780、mcf7和a549/ddp中的至少一种。
[0055]
以下将通过实施例对本发明进行详细描述。
[0056]
以下实施例中,在没有特别说明的情况下,所有商购获得的原料、试剂直接使用,不做进一步的处理。有机溶剂通过旋转蒸发器在减压下浓缩。
[0057]
以下过程用于说明青蒿琥酯衍生物i(n=6-12,r1为c1-c3的烷基)的制备
[0058][0059]
(1)将br(ch2)nbr(式iv所示的化合物)溶于乙腈中,自滴液漏斗向其中以10-15wt%/min的速度加入三苯基膦(式iii所示的化合物),化合物iv和iii的摩尔比为5:1,氮气保护下搅拌加热(70-90℃),反应20-25h。反应结束后,旋蒸除去溶剂,分别用乙酸乙酯和乙醚洗涤三次,柱层析纯化(二氯甲烷:甲醇=10-20:1)得到化合物v。
[0060]
(2)将化合物v和青蒿琥酯(式vi所示的化合物)溶于乙腈中,加入无水碳酸钾,化合物v、vi和无水碳酸钾的摩尔比为1:(1.2-1.6):(2-2.5),氮气保护下搅拌加热(70-85℃),反应6-12h。反应结束后,过滤,旋蒸除去溶剂,剩余物用二氯甲烷溶解,然后分别用饱和nahco3溶液和水洗涤2-3次,有机相干燥后柱层析纯化(二氯甲烷:甲醇=10-20:1)制备得到化合物i。
[0061]
以下过程用于说明青蒿琥酯衍生物ii(n=6-12,r1为c1-c3的烷基)的制备
[0062][0063]
(1)将br(ch2
)n
br(式iv所示的化合物)溶于乙腈中,自滴液漏斗向其中以10-15wt%/min的速度加入化合物vii,化合物iv和vii的摩尔比为5:1,氮气保护下搅拌加热至70-90℃,反应5-10h。反应结束后,旋蒸除去溶剂,分别用乙酸乙酯和乙醚洗涤三次,柱层析
纯化(二氯甲烷:甲醇=10-20:1)得到化合物viii。
[0064]
(2)将化合物viii和青蒿琥酯vi溶于乙腈中,加入无水碳酸钾,化合物viii、vi和无水碳酸钾的摩尔比为1:(1.2-1.6):(2-2.5),氮气保护下搅拌加热(70-85℃)进行取代反应,反应6-12h。反应结束后,过滤,旋蒸除去溶剂,剩余物用二氯甲烷溶解,然后分别用饱和nahco3溶液和水洗涤2-3次,有机相干燥后柱层析纯化(二氯甲烷:甲醇=10-20:1)制备得到化合物ii。
[0065]
为了更加直观,下面列举一些具体化合物的具体制备过程
[0066]
实施例1
[0067]
化合物1的合成:
[0068][0069]
向两口瓶中加入1,6-二溴己烷即化合物iv-1(1.154ml,7.5mmol)和15ml乙腈,氮气保护下搅拌加热至回流(82℃)。然后,自滴液漏斗向其中以15wt%/min的速度加入三苯基膦即化合物iii(393.44mg,1.5mmol)的乙腈(10ml)溶液。滴加完毕,氮气保护下回流(82℃)反应23h。反应结束后,旋蒸除去溶剂,分别用乙酸乙酯(10ml)和乙醚(10ml)洗涤三次,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=20:1),得到化合物v-1,淡黄色油状物(758mg,产率86%)。
[0070][0071]
向两口瓶中加入青蒿琥酯即化合物vi(308.34mg,0.802mmol)、化合物v-1(270.7mg,0.535mmol)、无水碳酸钾(184.76mg,1.337mmol)和15ml乙腈,氮气保护下回流(82℃)进行取代反应6.5h。反应结束后,过滤除去不溶物,旋蒸除去溶剂,剩余物用15ml二氯甲烷溶解,然后分别用饱和nahco3溶液(10ml)和水(10ml)洗涤2次。收集有机相,无水硫酸镁干燥,硅胶柱层析纯化(二氯甲烷:甲醇=15:1),得到化合物1,白色固体(357mg,产率83%,总产率71%)。
[0072]
实施例2
[0073]
化合物2的合成
[0074][0075]
参照实施例1的方法步骤,化合物iii(262.29mg,1.0mmol)和1,10-二溴癸烷即化合物iv-2(1.46ml,5.0mmol)在乙腈(25ml)中反应,得到化合物v-2,淡黄色油状物(461mg,产率82%)。接着,化合物v-2(431.92mg,0.768mmol)、vi(442.9mg,1.15mmol)和无水碳酸钾(212.29mg,1.54mmol)在乙腈(20ml)中进行取代反应,得到化合物2,白色固体(525mg,产率79%,总产率65%)。
[0076]
实施例3
[0077]
化合物3的合成
[0078][0079]
参照实施例1的方法步骤,化合物iii(262.29mg,1.0mmol)和1,12-二溴十二烷即化合物iv-3(1.176ml,5.0mmol)在乙腈(25ml)中反应,得到化合物v-3,淡黄色油状物(496mg,产率84%)。接着,化合物v-3(249.1mg,0.42mmol)、vi(283.7mg,0.74mmol)和无水碳酸钾(170mg,1.23mmol)在乙腈(20ml)中进行取代反应,得到化合物3,白色固体(277mg,产率74%,总产率62%)。
[0080]
实施例4
[0081]
化合物4的合成
[0082]
[0083]
向两口瓶中加入化合物iv-1(1.54ml,10mmol)和15ml乙腈,氮气保护下搅拌加热至70℃。然后,自滴液漏斗向其中以15wt%/min的速度加入1-甲基咪唑即化合物vii-1(159.4μl,2mmol)的乙腈(10ml)溶液。滴加完毕,氮气保护下70℃反应5h。反应结束后,旋蒸除去溶剂,分别用乙酸乙酯(10ml)和乙醚(10ml)洗涤三次,残留物用硅胶柱层析纯化(二氯甲烷:甲醇=10:1),得到化合物viii-1,黄色油状物(444mg,产率81%)。
[0084][0085]
向两口瓶中加入青蒿琥酯即化合物vi(232.6mg,0.61mmol)、化合物viii-1(164.4mg,0.50mmol)、无水碳酸钾(139.36mg,1.0mmol)和15ml乙腈,氮气保护下回流(82℃)进行取代反应6.5h。反应结束后,过滤除去不溶物,旋蒸除去溶剂,剩余物用15ml二氯甲烷溶解,然后分别用饱和nahco3溶液(10ml)和水(10ml)洗涤2次。收集有机相,无水硫酸镁干燥,硅胶柱层析纯化(二氯甲烷:甲醇=10:1),得到化合物4,淡黄色油状物(350mg,产率76%,总产率62%)。
[0086]
实施例5
[0087]
化合物5的合成
[0088][0089]
参照实施例4的方法步骤,化合物vii-1(159.4μl,2.0mmol)和iv-2(2.25ml,10.0mmol)在乙腈(25ml)中反应,得到化合物viii-2,淡黄色油状物(659mg,产率87%)。接着,化合物viii-2(530.2mg,1.39mmol)、vi(639.9mg,1.67mmol)和无水碳酸钾(383.5mg,2.77mmol)在乙腈(20ml)中进行取代反应,得到化合物5,淡黄色油状物(798mg,产率78%,总产率68%)。
[0090]
实施例6
[0091]
化合物6的合成
[0092][0093]
参照实施例4的方法步骤,化合物vii-1(159.4μl,2.0mmol)和iv-3(3.281mg,10.0mmol)在乙腈(25ml)中反应,得到化合物viii-3,淡黄色油状物(339mg,产率83%)。接着,化合物viii-3(530.2mg,0.78mmol)、vi(448.4mg,1.17mmol)和无水碳酸钾(214.9mg,1.55mmol)在乙腈(20ml)中反应,得到化合物6,淡黄色油状物(429mg,产率77%,总产率64%)。
[0094]
分别测试上述所得化合物的核磁数据(1h nmr)和质谱数据(hrms),将结果记于表1中。
[0095]
表1
[0096]
[0097]
[0098][0099]
测试例(抗肿瘤活性试验)
[0100]
对本发明的化合物进行肿瘤细胞增殖抑制试验,试验方法采用常规的mtt法。
[0101]
细胞株选用肝癌hepg2、宫颈癌hela、卵巢癌a2780、乳腺癌mcf7和顺铂耐药的肺癌a549/ddp,均购自中国科学院细胞库。培养液为dmem+10%fbs+双抗。
[0102]
样品液配制:待测化合物(上述实施例制备的化合物)用dmso(merck)溶解后,配成浓度为1mm的母液。用培养基稀释母液,配成药物的最终浓度分别为62.5μm、25μm、10μm、5μm、1μm和0.5μm。
[0103]
将抗肿瘤化合物顺铂(购自上海阿拉丁试剂有限公司)和青蒿琥酯(购自上海阿拉丁试剂有限公司)以同样的条件配成对照品溶液。
[0104]
96孔板每孔加入浓度为1.5
×
104个/ml的细胞悬液200μl,即3000个细胞/孔,置于37℃、5%co2培养箱内。细胞培养24小时贴壁后,弃掉上层培养液,加入含有样品的培养液
和对照品液,200μl/孔,37℃条件下作用48小时。每孔加入mtt 20μl,再加180μl培养基,置于培养箱内,作用4小时后弃掉培养基,加入200μl dmso,避光振摇10min,用multiskan fc型酶标仪测490nm od值,计算半数抑制浓度ic
50
。
[0105]
本发明中部分优选化合物的抗肿瘤活性详见表2,其中,化合物1-6是指相应实施例中制备的青蒿琥酯衍生物,如化合物1表示在实施例1中所得到的化合物,同理类推。
[0106]
表2本发明部分化合物对肿瘤细胞的半数抑制浓度ic
50
(μm)
[0107][0108][0109]
由表2的结果可知,本发明提供的化合物(青蒿琥酯衍生物)整体上表现出广谱、优异的抗肿瘤活性,对肝癌hepg2、宫颈癌hela、卵巢癌a2780、乳腺癌mcf7和顺铂耐药的肺癌a549/ddp均有优异的增殖抑制作用。部分化合物的抗肿瘤活性明显强于顺铂。例如,化合物3总体表现出最优的抗肿瘤活性,对宫颈癌hela和卵巢癌a2780的半数抑制浓度ic
50
都低于1μm,对顺铂耐药的肺癌a549/ddp的ic
50
低于2μm,对乳腺癌mcf7的ic
50
低于7μm。此外,部分化合物对顺铂耐药的a549/ddp肿瘤细胞仍表现出优异的抗肿瘤活性。又例如,化合物1、2、3和6对顺铂耐药的a549/ddp的ic
50
低于12μm,明显优于顺铂(ic
50
=44.8μm)。另外,本发明的大部分化合物的体外抗肿瘤活性明显优于青蒿琥酯。例如化合物1、2和6对宫颈癌hela的ic
50
分别为23.7、8.4和7.4μm,对顺铂耐药的a549/ddp的ic
50
分别为6.2、2.6和11.6μm。而青蒿琥酯对宫颈癌hela和顺铂耐药的a549/ddp的ic
50
分别为34.8和22.8μm。可见,本发明提供的化合物抗肿瘤活性明显较高。本发明提供的化合物具有全新的骨架结构,具有优异的抗肿瘤活性,可以进行抗肿瘤药物的开发。
[0110]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。