1.本发明属于电极材料领域,具体涉及一种酰胺化环状氮氧自由基化合物及其制备方法和应用。
背景技术:
2.为了解决可再生能源,如风能和太阳能的不稳定、不连续特性对电网造成冲击,亟需发掘灵活和可扩展的新型能源规模化存储技术。目前大规模电化学储能技术主要依靠无机类化合物为电极材料的全钒液流电池,考虑成本和环境危害性以及能量和功率密度,特别是长期使用含钒类金属资源,这对其发展应用提出了一定的限制作用,且面临资源与环境问题,急需开发新型的大规模液流电池储能技术。环状氮氧自由基化合物是一种含有稳定氮氧自由基结构的电极材料,其结构中稳定的氮氧自由基可以发生可逆的单电子氧化还原转化,在电池充放电循环中具有优异的倍率性能和循环稳定性,且属于一种资源丰富、环境友好和扩展性强的储能技术体系,其使用环状氮氧自由基化合物可作为电极材料的同时又具有无金属特点和使用廉价的阴离子交换膜甚至多孔透析膜作为氯化钾、氯化钠水溶液的优势。
3.目前报道的环状氮氧自由基化合物的种类较少,且环状氮氧自由基化合物的结构与物理化学性质及电化学性能之间的内在关系还不明晰。此外,现有环状氮氧自由基化合物正极材料的氧化还原电位低、水溶性低和电化学性能不稳定,导致环状氮氧自由基化合物基电池的能量和功率密度不高及循环稳定性不长。因此,研究环状氮氧自由基化合物结构与性能之间的构效关系,开发具有低成本、环境友好的高性能新型状氮氧自由基化合物作为新型水系有机液流电池电极材料具有重要意义。
技术实现要素:
4.本发明的目的在于克服上述现有技术的缺点,提供一种酰胺化环状氮氧自由基化合物及其制备方法和应用,以解决现有技术中已有的环状氮氧自由基化合物基电池的能量和功率不高,且循环稳定性不长的问题。
5.为达到上述目的,本发明采用以下技术方案予以实现:
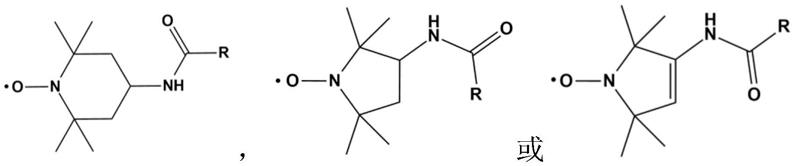
6.一种酰胺化环状氮氧自由基化合物,结构通式为:
[0007][0008]
其中,r为卤素基团、烷基链基团、含季铵盐阳离子基团、含磷酸盐阴离子基团、含磺酸盐阴离子基团中的一种或多种的组合。
[0009]
一种酰胺化环状氮氧自由基化合物的制备方法,包括以下步骤:
[0010]
步骤1,混合反应物a和反应物b,混合后加入二氯甲烷或吡啶,通过重复抽真空和充入惰性气体,使得反应体系处于无氧状态进行反应,反应后通过萃取、干燥、旋蒸获得红色固体;所述反应物a为哌啶氨、吡咯烷氨或吡咯啉氨;所述反应物b为卤代羧酸或酰氯;
[0011]
步骤2,在有机溶剂中加入红色固体和叔氨类、亚硫酸钠或亚磷酸三乙酯反应物,油浴回流后,将反应体系重复进行后处理若干次,获得酰胺化环状氮氧自由基化合物。
[0012]
本发明的进一步改进在于:
[0013]
优选的,步骤1中,所述反应物a和反应物b的混合摩尔比为1:(1~5)。
[0014]
优选的,步骤1中,二氯甲烷或吡啶的加入量为反应容器体积的1/3~1/2。
[0015]
优选的,步骤1中,反应温度为0~25℃,反应时间为6~18h。
[0016]
优选的,步骤2中,红色固体和叔氨类反应物的摩尔比为1:1~1:10。
[0017]
优选的,步骤2中,油浴回流温度为60~100℃,油浴回流时间为24~48h。
[0018]
优选的,步骤2中,有机溶剂为二氯甲烷、吡啶、丙酮、乙腈的一种或多种。
[0019]
一种述的酰胺化环状氮氧自由基化合物的应用,用于水系有机电池器件,所述水系有机电池器件由铜板集流体、蛇形流道石墨板、电极、正极电解液材料、离子交换膜、负极电解液材料、电极、蛇形流道石墨板、铜板集流体依次层叠组装构成;所述正极电解液材料为酰胺化环状氮氧自由基化合物。
[0020]
优选的,所述负极电解液材料由氯化锌、紫精类、蒽醌类、吩嗪类、喋啶类化合物制备而成;所述电极为高温退火处理或酸预处理后的石墨毡,或者是高温退火处理或酸预处理后的碳纸。
[0021]
与现有技术相比,本发明具有以下有益效果:
[0022]
本发明公开了一种酰胺化环状氮氧自由基化合物,该化合物通过调整氮氧自由基上接入官能团的结构,使得自由基化合物充分结合有机分子形成共轭结构,稳定性好,酰胺基团中氮上孤对电子占据的p轨道与羰基π*轨道的p-π共轭作用使其具有高旋转势垒和强化学惰性,进而使得碳氮单键具有双键的性质,加之可进一步引入水溶性的官能团,可有效提高酰胺化环状氮氧自由基电极材料的能量密度和循环稳定性,有效改善电极材料的氧化还原电位、水溶性和电化学稳定性能等性质。
[0023]
本发明还公开了一种酰胺化环状氮氧自由基化合物的制备方法,在制备所述酰胺化环状氮氧自由基化合物时,先将具有哌啶氨或吡咯烷氨或吡咯啉氨与卤代羧酸或酰氯进行脱水缩合反应制备出带有酰胺结构的环状自由基化合物,再加入含水溶性官能团化合物,混合加热发生取代反应,即可得到所述酰胺化环状自由基化合物,制备方法简单高效。
[0024]
本发明还公开了一种酰胺化环状氮氧自由基化合物的应用,能够应用于水系有机电池器件,具体的是应用于水系有机液流电池,以铜板为集流体,中性盐的水溶液为电解质;蛇形流道的石墨板作为正负极液的流场板;以石墨毡或碳纸作为反应电极,阴离子交换膜为隔膜,以还原态的吡咯啉/烷氮氧自由基及其衍生物作为正极材料电解液;以紫罗精衍生物或氯化锌中的一种作为负极材料电解液;电池器件整体性能优异、安全性高和成本低,在对可再生能源高效利用及存贮需求亟需的今天,有着广阔的发展应用和市场前景。
附图说明
[0025]
图1是本发明实施例一中固体产物的核磁共振氢谱;
[0026]
图2是本发明实施例二中固体产物的核磁共振氢谱;
[0027]
图3是本发明实施例三中固体产物的核磁共振氢谱;
[0028]
图4是本发明实施例六中正极材料电解液在不同扫速下的循环伏安曲线,扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒;
[0029]
图5是本发明实施例七中正极材料电解液在不同扫速下的循环伏安曲线,扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒;
[0030]
图6是本发明实施例八中正极材料电解液在不同扫速下的循环伏安曲线,扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒;
[0031]
图7是本发明实施例九中正极材料电解液在不同扫速下的循环伏安曲线,扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒;
[0032]
图8是本发明实施例十中正极材料电解液在不同扫速下的循环伏安曲线,扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒;
[0033]
图9是本发明实施例十一中所组装电池的充电和放电曲线,电流密度为10毫安/平方厘米;
[0034]
图10是本发明实施例十一中所组装电池的循环数-库伦效率-能量效率-充电容量-放电容量曲线,电流密度为10毫安/平方厘米;
[0035]
图11是本发明实施例十二中所组装电池的充电和放电曲线,电流密度为50毫安/平方厘米;
[0036]
图12是本发明实施例十二中所组装电池的循环数-库伦效率-能量效率-充电容量-放电容量曲线,电流密度为50毫安/平方厘米;
[0037]
图13是本发明实施例十三中所组装电池的充电和放电曲线,电流密度为50毫安/平方厘米;
[0038]
图14是本发明实施例十三中所组装电池的循环数-库伦效率-能量效率-充电容量-放电容量曲线,电流密度为50毫安/平方厘米;
具体实施方式
[0039]
本发明公开了一种酰胺化环状氮氧自由基化合物,其结构通式:
[0040][0041]
其中,r为卤素基团、烷基链基团、含季铵盐阳离子基团、含磷酸盐阴离子基团、含磺酸盐阴离子基团中的一种或多种的组合。
[0042]
一种酰胺化环状氮氧自由基化合物的制备方法,包括以下两个步骤:
[0043]
步骤一:
[0044]
在圆底烧瓶中加入混合反应物a和反应物b,反应物a为哌啶氨、吡咯烷氨或吡咯啉氨,反应物b为卤代羧酸或酰氯,其中反应物a和反应物b的摩尔比为1:1~1:5;在圆底烧瓶反应器中,加入二氯甲烷或吡啶作为有机溶剂,加入量为反应容器体积的1/3~1/2,通过三
次抽真空-充入惰性气体操作使反应体系处于无氧状态,在~25℃下反应进行6~18h,具体的,反应物a和反应物b发生的酰胺化反应为脱水缩合反应;反应后通过去离子水萃取,无水硫酸镁干燥,旋蒸得到红色固体。对于生成目标产物的r基为卤素或烷基链的酰胺化环状氮氧自由基化合物,步骤一即生成了最终的产物;若r基为含季铵盐阳离子、磷酸盐阴离子或磺酸盐阴离子基团,继续进行步骤二。
[0045]
步骤二:
[0046]
将红色固体置入圆底烧杯中,加入有机溶剂和叔氨类反应物,有机溶剂加入量为反应容器体积的1/3~1/2,红色固体和叔氨类反应物的摩尔比为1:1~1:10,有机溶剂为二氯甲烷、吡啶、丙酮、乙腈的一种或多种,然后在60~100℃油浴下回流24~48h,过滤,旋蒸,干燥,重复操作三次,得到红色固体产物。
[0047]
所述的水系有机电池器件由铜板集流体、蛇形流道石墨板、电极、正极电解液材料、离子交换膜、负极电解液材料、电极、蛇形流道石墨板、铜板集流体依次层叠组装构成,所述的正极电解液材料由酰胺化环状氮氧自由基化合物制备而成。所述的负极电解液材料由以水溶性高的氯化锌或紫精类或蒽醌类或吩嗪类或喋啶类化合物制备而成。所述的电解质为氯化钾、氯化钠、硝酸钾、硝酸钠水溶液中的一种或者多种的任意组合。所述的电极为高温退火处理或者酸预处理后的石墨毡或碳纸。
[0048]
实施例1
[0049]
向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和乙酸(28mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后25℃搅拌12h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到红色固体产物4-乙酰氨基-2,2,6,6-四甲基哌啶氮氧化物。核磁共振氢谱表征结果如图1所示。1h nmr(600mhz,d2o,25℃):δ3.97(t,1h),1.84(s,3h),1.73(m,4h),1.34(m,2h),1.09(d,12h)。
[0050]
实施例2
[0051]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(28mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在25℃搅拌12h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0052]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),三甲胺(12mmol)。80℃油浴条件下回流36h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[3-三甲铵乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。核磁共振氢谱表征结果如图2所示。1h nmr(600mhz,dmso-d6,25℃):δ5.18(t,1h),3.69(t,2h),2.68(t,2h),1.95(m,4h),1.51(m,2h),1.19(d,12h)。
[0053]
实施例3
[0054]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(28mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后常温搅拌12h。反应
结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0055]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),n-甲基咪唑(12mmol)。80℃油浴条件下回流36h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[甲基咪唑盐乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。核磁共振氢谱表征结果如图3所示。1hnmr(600mhz,d2o,25℃):δ7.65(s,2h),7.53(s,2h),4.88(s,2h),4.07(t,1h),3.82(s,3h),1.84(d,2h),1.42(t,2h),1.12(d,12h)。
[0056]
实施例4
[0057]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(28mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在0℃搅拌18h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0058]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),亚硫酸钠(12mmol)。80℃油浴条件下回流45h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[3-磺酸钠乙酰胺]-2,2,6,6-四甲基哌啶氮氧化物。
[0059]
实施例5
[0060]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(100mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在0℃搅拌18h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0061]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),亚磷酸三乙酯(16mmol)。80℃油浴条件下回流45h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[3-磷酸酸钠乙酰胺]-2,2,6,6-四甲基哌啶氮氧化物。
[0062]
以上制备方法中所用原料及溶剂均从其它厂家购买。
[0063]
实施例6
[0064]
将0.02mmol 4-乙酰氨基-2,2,6,6-四甲基哌啶氮氧化物加入到10ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备2mm的正极材料电解质溶液。将其用三电极体系进行循环伏安测试,所用参比电极为银/氯化银电极,工作电极为玻璃碳电极,对电极为铂片电极。所选扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒,得到图4所示循环伏安曲线,氧化还原可逆性较好,且平均电位在0.63v左右。
[0065]
实施例7
[0066]
将0.02mmol 4-[3-三甲铵乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物加入到10ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备2mm的正极材料电解质溶液。将其用三电极体系进行循环伏安测试,所用参比电极为银/氯化银电极,工作电极为玻璃碳电极,对电极为铂片电极。所选扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒,得到图5所示循环伏安曲线氧化还原可逆性较好,且平均电位在0.63v左右。
[0067]
实施例8
[0068]
将0.02mmol 4-[甲基咪唑盐乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物加入到10ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备2mm的正极材料电解质溶液。将其用三电极体系进行循环伏安测试,所用参比电极为银/氯化银电极,工作电极为玻璃碳电极,对电极为铂片电极。所选扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒,得到图6所示循环伏安曲线,氧化还原可逆性较好,且平均电位在0.63v左右。
[0069]
实施例9
[0070]
将0.02mmol 4-[磺酸钠乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物加入到10ml1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备2mm的正极材料电解质溶液。将其用三电极体系进行循环伏安测试,所用参比电极为银/氯化银电极,工作电极为玻璃碳电极,对电极为铂片电极。所选扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒,得到图7所示循环伏安曲线,氧化还原可逆性较好,且平均电位在0.63v左右。
[0071]
实施例10
[0072]
将0.02mmol 4-[磷酸钠乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物加入到10ml1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备2mm的正极材料电解质溶液。将其用三电极体系进行循环伏安测试,所用参比电极为银/氯化银电极,工作电极为玻璃碳电极,对电极为铂片电极。所选扫速为25毫伏/秒,50毫伏/秒,100毫伏/秒,250毫伏/秒和500毫伏/秒,得到图8所示循环伏安曲线,氧化还原可逆性较好,且平均电位在0.63v左右。
[0073]
实施例11
[0074]
称取0.7mmol 4-乙酰氨基-2,2,6,6-四甲基哌啶氮氧化物加入到7ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备0.1m的正极材料电解质溶液;称取3mmol氯化锌溶解到10ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备0.3m氯化锌溶液作为负极材料电解液。将上述正极材料电解液、负极材料电解液分别存储于正负极储液罐内,用蠕动泵循环。正负极材料电解液通过管道各自循环,交汇于隔膜两侧,在隔膜两侧的石墨毡或碳纸电极发生氧化还原反应,正负两电极连通于电源负载,电路传递电子,隔膜传递正负离子,形成回路。对该电池进行充放电性能和循环稳定性评估,采用10毫安/平方厘米的电流密度进行恒流充放电测试。可以看到电池充放电曲线表现出正常的充电和放电平台,且首次库伦效率在99%以上,图9所示。其在100圈的连续循环内,容量衰减率仅为0.01%/圈,库伦效率平均保持在99.5%以上,能量效率保持在70%左右,循环性能较好,图10所示。
[0075]
实施例12
[0076]
称取0.7mmol 4-[3-三甲铵乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物加入到7ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备0.1m的正极材料电解质溶液;称取1mmol紫精衍生物1,1'-双[3-(三甲基铵)丙基]-4,4'-联吡啶四氯化物溶解到10ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备0.1m紫精衍生物溶液作为负极材料电解液。将上述正极材料电解液、负极材料电解液分别存储于正负极储液罐内,用蠕动泵循环。正负极材料电解液通过管道各自循环,交汇于隔膜两侧,在隔膜两侧的石墨毡或碳纸电极发生氧化还原反应,正负两电极连通于电源负载,电路传递电子,隔膜传递正负离子,形成回路。对该电池进行充放电性能和循环稳定性评估,采用50毫安/平方厘米的电流密度进行恒流充放电测试。可以看到电池充放电曲线表现出正常的充电和放电平台,且首次库伦效率在99%以上,图11所
示。其在500圈的连续循环内,容量衰减率仅为0.005%/圈,库伦效率平均保持在99.7%以上,能量效率保持在66%左右,循环性能优异,图12所示。
[0077]
实施例13
[0078]
称取0.8mmol 4-[甲基咪唑盐乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物加入到8ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备0.1m的正极材料电解质溶液;称取36mmol氯化锌溶解到12ml 1m的氯化钾溶液中,超声0.5h,搅拌0.5h,制备0.3m氯化锌溶液作为负极材料电解液。将上述正极材料电解液、负极材料电解液分别存储于正负极储液罐内,用蠕动泵循环。正负极材料电解液通过管道各自循环,交汇于隔膜两侧,在隔膜两侧的石墨毡或碳纸电极发生氧化还原反应,正负两电极连通于电源负载,电路传递电子,隔膜传递正负离子,形成回路。对该电池进行充放电性能和循环稳定性评估,采用50毫安/平方厘米的电流密度进行恒流充放电测试。可以看到电池充放电曲线表现出正常的充电和放电平台,且首次库伦效率在99%以上,图13所示。其在500圈的连续循环内,容量衰减率仅为0.003%/圈,库伦效率平均保持在99.6%以上,能量效率保持在69%左右,循环性能优异,图14所示。
[0079]
实施例14
[0080]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(25mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在12℃搅拌14h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0081]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),三甲胺(8mmol)。90℃油浴条件下回流30h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[3-三甲铵乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。
[0082]
实施例15
[0083]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(75mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在20℃搅拌6h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0084]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),n-甲基咪唑(80mmol)。70℃油浴条件下回流40h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[甲基咪唑盐乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。
[0085]
实施例16
[0086]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(125mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在10℃搅拌8h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0087]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),三甲胺(24mmol)。100℃油浴条件下回流24h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[3-三甲铵乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。
[0088]
实施例17
[0089]
(1)向装有60ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(50mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在8℃搅拌10h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0090]
(2)向装有60ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),n-甲基咪唑(40mmol)。60℃油浴条件下回流48h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[甲基咪唑盐乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。
[0091]
实施例18
[0092]
(1)向装有50ml干燥二氯甲烷的100ml圆底烧瓶中依次加入4-氨基-2,2,6,6-四甲基哌啶氮氧化物(25mmol)和氯乙酸(100mmol)。通过三次抽真空-充入惰性气体操作使反应体系处于无氧状态,冰水浴搅拌下缓慢滴加吡啶(28mmol),滴加完毕后在0℃搅拌18h。反应结束后,依次用去离子水(30ml)、碳酸氢钠(10wt.%)、盐酸溶液(2m)、去离子水(30ml)萃取三次得有机相。用无水硫酸镁干燥,旋蒸得到固体中间体。
[0093]
(2)向装有50ml乙腈的100ml圆底烧瓶中依次加入红色固体中间体(8mmol),三甲胺(64mmol)。80℃油浴条件下回流45h后,旋蒸除去溶剂得到淡红色固体,用丙酮洗涤多次,过滤干燥得淡红色固体产物4-[3-三甲铵乙酰胺氯]-2,2,6,6-四甲基哌啶氮氧化物。
[0094]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。