一种含有人pd1基因sgrna的t细胞及其应用
技术领域
1.本发明涉及一种含有人pd1基因sgrna的t细胞及其应用,属于基因工程技术领域。
背景技术:
2.pd-1(程序性死亡受体1,programmed death 1) 蛋白属于免疫球蛋白超家族成员,可表达于活化的t细胞、b细胞和骨髓细胞,以及cd4-cd8-胸腺细胞。pd-1有两个配体,即pd-l1(b7-h1)和pd-l2(b7-dc),均为b7家族中的新成员。pd-1是免疫抑制性受体,与其配体pd-l1、pd-l2相互作用传递抑制性信号,在免疫应答中发挥负向调控作用。t细胞上的pd-1与肿瘤细胞中的pd-l1/ pd-l2的结合,可抑制活化的t细胞攻击肿瘤细胞,导致免疫系统不能发挥全部作用,使肿瘤细胞逃逸。
3.crispr/cas9技术是非常热门的基因编辑操作工具,目前华西医院已报道利用crispr技术敲除t细胞中的pd1,具有安全无副作用的优势。目前用于设计sgrna的网站有十多个,同一个基因设计出来的sgrna序列多达数百个,其每个sgrna导入细胞内后造成的敲除效率是不同的,需要筛选出敲除效率高的sgrna。
4.cn103820454b专利虽然提供了180个pd1的sgrna序列,但只给出了其中4个单sgrna和4个双sgrna的效果,对于其余sgrna并没有验证其敲除效率。cn107586777a专利虽然也提供了131个sgrna,但对于其效果只提供了sgrna活性,没有涉及具体的敲除效率。
5.cn105671083b专利仅公开了一个sgrna序列的效果,其余sgrna序列具有的敲除效率是未知的。
技术实现要素:
6.针对现有技术存在的不足,本发明提供一种含有人pd1基因sgrna的t细胞,实现以下发明目的:筛选出高敲除率的sgrna,提高t细胞对肿瘤细胞的杀伤力。
7.为解决上述技术问题,本发明采取以下技术方案:一种含有人pd1基因sgrna的t细胞,所述sgrna是敲除pd1基因的靶点;所述sgrna的核苷酸序列为序列表中seq id no:4、seq id no:6或seq id no:8所示;将含有sgrna的质粒转染到t细胞中,得到含sgrna的crispr-pd1-t细胞。
8.以下是对上述技术方案的进一步改进:所述crispr-pd1-t细胞的制备方法如下:步骤1、采集结肠癌患者外周血,分离出单个核细胞,将收集的单个核细胞用生理盐水清洗后,得到t细胞;收集的血浆经灭活、离心后,取上清,得到血浆上清。
9.步骤2、将t细胞加入kbm551培养基重悬细胞,使细胞密度保持在1
×
106个/ml,添加γ干扰素浓度为200 iu/ml,放入培养箱培养。
10.步骤3、γ干扰素作用24h后,添加cd3/cd28单抗和il-2,浓度分别为50ng/ml和300 iu/ml,培养48h后得到活化的t细胞。
11.步骤4、在6孔板中加入2ml/孔新鲜的含有300 iu/ml il-2的kbm551培养基,置于37℃培养箱中预热,得到预热的培养基;收集活化的t细胞,取7.5
×
107个细胞,用1.5ml电转液重悬,加入30μg含有sgrna的质粒混合均匀,选择电活化t细胞程序电转,电转完成后将细胞立即转移至预热的培养基中,放回37℃培养箱中继续培养。
12.步骤5、48h后,取细胞进行流式检测,检测电转的效率;取样计数,根据计数结果补充相应的免疫细胞培养基和il-2,使细胞的浓度维持在1
×
106个/ml,il-2的浓度维持在300iu/ml,添加5vol%的血浆上清。
13.步骤6、48h后进行细胞计数,根据计数结果补充相应的免疫细胞培养基和il-2,使细胞的浓度维持在1
×
106个/ml,il-2的浓度维持在300iu/ml,补加5vol%的血浆上清。
14.步骤7、48h后收集细胞,得到crispr-pd1-t细胞。
15.所述含有人pd1基因sgrna的t细胞在治疗抗肿瘤药物中的应用。
16.与现有技术相比,本发明取得以下有益效果:(1)本发明所述针对人pd1基因的sgrna靶点,转染到稳定表达pd1的293t细胞中,对pd1基因的敲除率为71.92-90.97%;转染到t细胞中,对pd1基因的敲除率为58.26-72.67%。
17.(2)本发明所述含人pd1基因的sgrna靶点的t细胞(crispr-pd1-t细胞),对结肠癌细胞系lovo均表现出显著的特异性细胞毒性作用,效靶比为3:1时,crispr-pd1-t细胞对结肠癌lovo细胞系的杀伤效率为68.17-83.76%;效靶比为10:1时,crispr-pd1-t细胞,对结肠癌lovo细胞系的杀伤效率为77.36-95.13%;并呈现效靶比梯度依赖性即效靶比越高细胞毒性作用越强。
18.(3)本发明所述含人pd1基因的sgrna靶点的t细胞(crispr-pd1-t细胞)对宫颈癌hela细胞系具有特异性细胞毒性作用。本发明筛选制备的三种crispr-pd1-t细胞对宫颈癌hela细胞系均表现出显著的特异性细胞毒性作用,效靶比为3:1时,对宫颈癌hela细胞系的杀伤效率为68.67-82.73%;效靶比为10:1时,对宫颈癌hela细胞系的杀伤效率为77.1-93.86%。
19.(4)本发明所述含人pd1基因的sgrna靶点的t细胞(crispr-pd1-t细胞)对blab/c小鼠肿瘤生长具有抑制作用,从第一次免疫起10天内,65.3-80%小鼠几乎触摸不到肿瘤;第一次免疫后10d,小鼠皮下肿瘤组织的肿瘤均值为59-330 mm3,第一次免疫后12d,小鼠皮下肿瘤组织的肿瘤均值为29-290 mm3,第一次免疫后15d,小鼠皮下肿瘤组织的肿瘤均值为7-225mm3。
附图说明
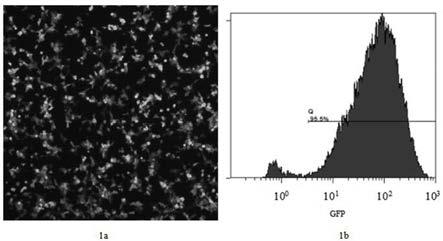
20.图1为293t-pd1稳转细胞系的免疫荧光和流式图;其中1a为293t-pd1稳转细胞系的免疫荧光图,1b为293t-pd1稳转细胞系的流式图;图2为不同sgrna转染293t-pd1稳转细胞系的免疫荧光图;2a为sg1转染293t-pd1稳转细胞系的免疫荧光图;2b为sg2转染293t-pd1稳转细胞系的免疫荧光图;2c为sg3转染293t-pd1稳转细胞系的免疫荧光图;
2d为sg4转染293t-pd1稳转细胞系的免疫荧光图;2e为sg5转染293t-pd1稳转细胞系的免疫荧光图;2f为sg6转染293t-pd1稳转细胞系的免疫荧光图;2g为sg7转染293t-pd1稳转细胞系的免疫荧光图;2h为sg8转染293t-pd1稳转细胞系的免疫荧光图;图3 为不同sgrna转染293t-pd1稳转细胞系的t7en1酶切图;图4为本发明中crispr-pd1-t细胞对结肠癌细胞系lovo杀伤率图;图5为本发明中crispr-pd1-t细胞对宫颈癌细胞系hela杀伤率图;图6为本发明中实施例7免疫后不同天数小鼠皮下肿瘤组织均值的折线图。
具体实施方式
21.实施例1:针对人pd1基因crispr/cas9载体的构建1、筛选针对人pd1基因的有效sgrna靶点根据pd1的基因序列,设计针对pd1基因的有效的sgrna靶点。
22.表1列出了其中8条针对pd1基因的有效sgrna靶点序列。
23.表1 靶点pd1基因的sgrna靶点序列 2、重组px458载体的构建本发明中选择的载体为px458载体(购自addgene),利用常规方法将表1中每个sgrna序列分别构建到px458载体中,其重组载体名称按顺序从上到下为px458-pd1-sg1、px458-pd1-sg2、px458-pd1-sg3、px458-pd1-sg4、px458-pd1-sg5、px458-pd1-sg6、px458-pd1-sg7、px458-pd1-sg8。
24.本发明中,8个重组质粒采用氯化铯法进行质粒提纯与浓缩,使其浓度达到1
µ
g/
µ
l。
25.实施例2:构建稳定表达pd1基因的293t细胞1、构建表达pd1蛋白的慢病毒本发明中选择的慢病毒载体为cd513b(购自addgene,pcdh-cmv-nluc-ef1α-coprfp-t2a-puro),将pd1基因编码区(genbank:ay238517.1)全长序列委托北京博迈德基因技术有限公司合成全长序列,利用xbai和ecori分别双酶切pd1基因和cd513b载体,回收酶切产物,利用t4 dna将两者连接起来,形成含有pd1基因的cd513b载体,命名为
cd513b-pd1,进行测序验证,正确后提取质粒,去除内毒素,本发明中提取的cd513b-pd1质粒浓度为600ng/
µ
l。
26.2、利用293t细胞包装慢病毒复苏293t细胞,传代3次后用于转染;六孔板中按6
×
105个细胞/孔进行接种,加入2ml 含有10vol% fbs的dmem培养基(均购自gibco公司),为第二天的转染做准备。
27.转染前将六孔板中的培养基换成新的dmem培养基,37℃温箱中培养1h;重组慢病毒质粒cd513b-pd1、与包装质粒pspax2与pmd2.g按4:2:3质量比例混合后,加入60
µ
l fugene hd(购自promega公司)转染试剂转染293t细胞,48小时后,显微镜下观察293t细胞转染后的形态变化和gfp的表达;72h后将含有病毒的细胞培养上清收集到离心管中,3500rpm/min离心10min,去除细胞碎片,4.5μm滤器过滤后以70000g,4℃离心2h,然后用100
µ
l的pbs重悬,得到病毒悬液,分装后放入-80℃保存;病毒悬液中重组慢病毒cd513b-pd1的病毒滴度为3.2
×
10
8 tu/ml。
28.3、慢病毒感染293t细胞,并用嘌呤霉素筛选稳定表达pd1的293t细胞六孔板内将复苏后293t细胞以6
×
105个/孔的密度铺板,加入2ml 含有10vol% fbs的dmem培养基孵育过夜,去除旧的培养基,加入moi=5的含重组慢病毒cd513b-pd1的病毒悬液(步骤2制备的病毒悬液),并加入1.5ml的无血清dmem培养基;病毒转导后6h,再添加1ml含10vol% fbs的dmem培养基到细胞培养板内,然后孵育过夜;病毒转导后48h,更换2ml新鲜的筛选培养基,继续培养;每2天替换新鲜配制的筛选培养基,每天检测细胞并观察活细胞生长比例,以及gfp表达的水平及所占比例;当gfp表达比率达到95%以上时(见图1),筛选成功,命名为293t-pd1,用pd1流式抗体来检测筛选后的293t细胞中pd1的表达率;本发明中,pd1的表达率为95.5%(见图1)。
29.上述筛选培养基为含有2
µ
g/ml的嘌呤霉素、10vol% fbs的dmem培养基。
30.实施例3:pd1 sgrna敲除效率的测定将293t-pd1细胞以1
×
106个/孔的密度接种在六孔板内,孵育过夜。
31.将构建的8个不同的含pd1 sgrna的重组px458载体分别取4μg,与10
µ
l fugene hd转染试剂混合,加入dmem培养基至100
µ
l,静置15min形成复合物,将六孔板内的培养基吸掉,加入静置好的复合物,添加dmem培养基至2ml,放在37℃、5%co2的培养箱内培养6h,更换含有10vol%fbs的dmem培养基继续培养48h,荧光显微镜下观察转染情况(见图2),收集每组1孔细胞,利用flag流式抗体进行转染效率的测定;同时其余两孔细胞进行传代,传代后3天收集各组细胞,利用pd1流式抗体进行检测。
32.实验中设置两组对照组,对照组1是相同条件下培养的293t-pd1,对照组2是以空载px458转染293t-pd1;每组实验组设3个复孔,取其平均值。
33.本发明中,每组的转染率和pd1的表达效率如表2;根据敲除率公式,计算每组的敲除率;结果显示293t-pd1-px458-sg4的敲除率最高,达到90.97%;293t-pd1-px458-sg6的敲除率为71.92%;293t-pd1-px458-sg8的敲除率为84.78%。
34.表2 实验组和对照组转染率、pd1表达率和敲除率
实施例4:利用t7en1酶切验证(1)将实施例3收集的各组细胞利用博迈德的磁珠法基因组提取试剂盒提取dna,用乙醇沉淀法纯化获得的pcr回收产物,利用表3中的引物扩增目的片段,其中sg5和sg7利用pd1-f2/r2引物扩增,其余的用pd1-f1/r1引物扩增。
35.(2)对于纯化的基因组dna的pcr产物,配制以下反应体系:dna(100ng/
µ
l)500ng;10
×
t7enbuffer2
µ
l;无核酸酶ddh2o加至19
µ
l;总体积19
µ
l;将反应体系混合,高速离心数秒,在95℃加热5分钟;退火,将解链后的pcr产物冷却至室温。
36.(3)每反应体系加入1
µ
l浓度为2u/
µ
l的t7核酸内切酶i,在37℃反应40分钟。
37.(4)在每个酶切反应体系中加入2μl的10
×
loadingbuffer并混匀;将一半的反应体系混合物加到含有2%琼脂糖胶的孔中,在tae或tbe缓冲液中进行电泳。
38.结果见图3,sg1到sg8均有两条目的条带,其中sg4目的条带最亮(跑胶图中下面两条带是目的条带),与其敲除率相对应。表4为pcr扩增长度和相对应的酶切片段长度。
39.表3扩增目的片段的引物表4t7en1酶切检测目的片段
实施例5crispr-pd1-t细胞的制备本实施例中对293t-pd1初筛得到的敲除率高的sg4、sg6、sg8进行验证,利用同一份外周血分离培养的免疫细胞进行电转,本次实验分为对照组和实验组,对照组有两组,一组为利用px458空载电转,一组为不进行电转的正常培养,实验组有三组,分别为px458-pd1-sg4、px458-pd1-sg6、px458-pd1-sg8。
40.1、采集结肠癌患者外周血,利用淋巴细胞分离液(购自天津灏洋生物制品科技有限责任公司)分离出单个核细胞,将收集的单个核细胞用生理盐水清洗2遍,得到t细胞;收集的血浆56℃灭活30min后,3000rpm离心20min,取上清放置于4℃冰箱备用。
41.2、根据t细胞的计数结果加入kbm551培养基(购自corning)重悬细胞,使细胞密度保持在1
×
106个/ml,添加γ干扰素浓度为200iu/ml,放入培养箱培养。
42.3、γ干扰素作用24h后,添加cd3/cd28单抗(购自北京同立海源生物科技有限公司)和il-2(购自江苏金丝利药业股份有限公司),浓度分别为50ng/ml和300iu/ml,培养48h后,得到活化的t细胞培养液。
43.4、电转前准备工作,在6孔板中加入2ml/孔新鲜的含有300iu/mlil-2的kbm551培养基,置于37℃培养箱中预热;将电转液(购自lonza的p3primarycell4dxkitl核转试剂盒,v4xp-3024)从4℃冰箱中拿出,取1.5ml电转液置于离心管中恢复至室温,剩余的电转液放回4℃冰箱;收集活化的t细胞,取7.5
×
107个细胞,用1.5ml电转液重悬,平均分为五份,其中一份直接添加到预热的6孔板中,剩余4份分别加入6μg的px458、px458-pd1-sg4、px458-pd1-sg6、px458-pd1-sg8混合均匀,每个电转杯中加入的t细胞数量为5
×
106个,每份需要3个电转杯。
44.将电转杯放入lonza4d电转仪中,选择电活化t细胞程序电转,电转完成后将细胞立即转移至预热的培养基中,放回37℃培养箱中继续培养。
45.5、48h后,取细胞进行流式检测,检测电转的效率;取样计数,根据计数结果补充相应的免疫细胞培养基和il-2,使细胞的浓度维持在1
×
106个/ml,il-2的浓度维持在300iu/
ml,添加5vol%的血浆上清。
46.6、48h后进行细胞计数,根据计数结果补充相应的免疫细胞培养基和il-2,使细胞的浓度维持在1
×
106个/ml,il-2的浓度维持在300iu/ml,补加5vol%的血浆上清。
47.7、48h后收集细胞,对细胞进行计数、活率检测、支原体检测,利用流式细胞仪对各组细胞进行pd1蛋白表达检测,计算实验组的敲除率。
48.本发明中,pd1蛋白的表达率如表5所示,其中px458-pd1-sg4-t的敲除率最高,为72.67%,px458-pd1-sg6-t和px458-pd1-sg8-t的敲除率相近,分别为58.26%、60%。
49.表5 各组实验中pd1蛋白的表达率实施例6 crispr-pd1-t细胞对结肠癌lovo细胞系的杀伤活性研究体外毒性实验使用的材料如下:以结肠癌细胞系lovo作为靶细胞,效应细胞为如实施例5所制备的体外培养的crispr-pd1-t细胞,分别为px458-pd1-sg4-t、px458-pd1-sg6-t、px458-pd1-sg8-t、px458-t、t细胞。效靶比视情况分别为10:1、3:1、1:1和1:3,靶细胞数量为1
×
104个/孔,根据不同效靶比对应效应细胞。各组均设3个复孔,取3个复孔的平均值。检测时间为细胞混合后4 h。其中各实验组和各对照组如下:各实验组:各靶细胞+表达不同嵌合抗原受体的t细胞,对照组1:靶细胞最大释放ldh,需加入一定体积的细胞裂解液 ;对照组2:靶细胞自发释放ldh;对照组3:效应细胞自发释放ldh;对照组4:空白培养基的背景;对照组5:体积校准的背景 ,空白培养基加入一定体积的细胞裂解液。
50.检测方法:采用cytotox96
®
非放射性细胞毒性检测试剂盒(promega公司)检测效应细胞对靶细胞的杀伤效率。该方法是基于比色法的检测方法,可替代51cr释放法。cytotox检测定量地测量乳酸脱氢酶(ldh)。ldh是一种稳定的胞质酶,在细胞裂解时会释放出来,其释放方式与51cr在放射性分析中的释放方式基本相同。释放出的ldh培养基上清中,可通过30分钟偶联的酶反应来检测,在酶反应中ldh可使一种四唑盐(int)转化为红色的甲臜(formazan),生成的红色产物的量与裂解的细胞数成正比(具体参照cytotox96
®
非
放射性细胞毒性检测试剂盒说明书)。细胞毒性计算公式为:实验结果表明:具体如图4和表6所示,本发明筛选制备的三种crispr-pd1-t细胞对结肠癌细胞系lovo均表现出显著的特异性细胞毒性作用,其中px458-pd1-sg4-t的细胞毒性作用要高于px458-pd1-sg6-t和px458-pd1-sg8-t的细胞毒性作用,效靶比为3:1时,对结肠癌lovo细胞系的杀伤效率为68.17-83.76%;效靶比为10:1时,对结肠癌lovo细胞系的杀伤效率为77.36-95.13%;并呈现效靶比梯度依赖性即效靶比越高细胞毒性作用越强;其中px458-pd1-sg4-t在效靶比10:1时对结肠癌细胞lovo的细胞毒性高达95.13%。效靶比依赖性的数据进一步显示本发明的crispr-pd1-t细胞对结肠癌细胞lovo的特异性细胞毒性作用。表6crispr-pd1-t细胞对结肠癌lovo细胞系的杀伤效率实施例7:crispr-pd1-t细胞对宫颈癌hela细胞系的杀伤活性研究根据实施例6的实验方法验证crispr-pd1-t细胞对宫颈癌hela细胞系的体外杀伤活性,结果如图5和表7所示,本发明筛选制备的三种crispr-pd1-t细胞对宫颈癌hela细胞系均表现出显著的特异性细胞毒性作用,效靶比为3:1时,对宫颈癌hela细胞系的杀伤效率为68.67-82.73%;效靶比为10:1时,对宫颈癌hela细胞系的杀伤效率为77.1-93.86%;并呈现效靶比梯度依赖性即效靶比越高细胞毒性作用越强;其中px458-pd1-sg4-t在效靶比10:1时对宫颈癌hela细胞的细胞毒性高达93.86%;px458-pd1-sg4-t的细胞毒性作用要高于px458-pd1-sg6-t和px458-pd1-sg8-t的细胞毒性作用。
51.效靶比依赖性的数据进一步显示本发明的crispr-pd1-t细胞对宫颈癌hela细胞系的特异性细胞毒性作用。
52.表7crispr-pd1-t细胞对宫颈癌hela细胞系的杀伤效率
实施例8:crispr-pd1-t细胞对blab/c小鼠肿瘤生长抑制作用18-22g雄性blab/c小鼠(购自沈阳蓝谱达斯实验用品科技有限公司)于动物房饲养(室温23
±
2℃,湿度50%
±
10%),收集对数期的结肠癌lovo细胞,磷酸盐缓冲液(pbs)稀释至2
×
105个/ml。无菌条件下,小鼠左腋下接种0.2ml结肠癌lovo细胞悬浮液,观察3-5d,待腋下出现米粒大小较硬的结节作为建模成功的标准。
53.blab/c结肠癌模型小鼠(游标卡尺量取皮下肿瘤组织块的大小为90-100mm3)随机分成6组,每组20只,开始注射治疗实验。
54.实验组分别为:a.对照组,尾部静脉注射同等体积的生理盐水;b.治疗一组,尾部静脉注射2
×
106个细胞/只正常t细胞;c.治疗二组,尾部静脉注射2
×
106个细胞/只px458空载电转的t细胞;d.治疗三组,尾部静脉注射2
×
106个细胞/只px458-pd1-sg4-t细胞;e.治疗四组,尾部静脉注射2
×
106个细胞/只px458-pd1-sg6-t细胞;f.治疗五组,尾部静脉注射2
×
106个细胞/只px458-pd1-sg8-t细胞。
55.每周免疫上述各组小鼠一次,连续免疫两周,每天通过游标卡尺量取各个实验组小鼠皮下肿瘤组织块大小,并记录,用肿块均值绘制肿瘤生长曲线图,结果如图6和表8所示。
56.表8免疫后不同天数小鼠皮下肿瘤组织的肿瘤均值图6和表8结果显示,治疗三、四、五组可以明显减少肿瘤组织的大小,第一次免疫
后10d,小鼠皮下肿瘤组织的肿瘤均值为59-330 mm3,第一次免疫后12d,小鼠皮下肿瘤组织的肿瘤均值为29-290 mm3,第一次免疫后15d,小鼠皮下肿瘤组织的肿瘤均值为7-225mm3。