1.本发明属于精细化工领域,具体涉及合成四缩醛的方法。
背景技术:
2.四缩醛是用于合成2,7-二甲基-2,4,6-辛三烯-1,8-二醛的一种重要中间体。
3.2,7-二甲基-2,4,6-辛三烯-1,8-二醛结构式为是合成类胡萝卜素产品的必要中间体,对于β-胡萝卜素、角黄素、虾青素、叶黄素、番茄红素等物质的合成意义重大。
4.类胡萝卜素作为一类重要的天然色素的总称。在饲料助剂、食品着色剂、营养强化剂、医药及日化领域具有广泛的应用和较高的市场附加值。
5.苏联文献j.gen.chem.ussr,34,第64页(1964),上述ii和iii化合物(r1=r2=ch3)的烯醇醚缩合反应在zncl2+bf3醚合物存在但不添加溶剂的条件下进行。四缩醛通过蒸馏以71%的收率分离,该方法制备的目标产物四缩醛收率较低,不利于大规模工业化生产。
6.wo2005077874提供了一种改进的制备2,7-二甲基辛-2,4,6-三烯二醛的方法,其中,丁烯二醛双缩醛与甲基-1-丙烯基醚等的烯醇醚在无水氯化铁等路易斯酸催化剂存在下进行双烯醇醚缩合以生成四缩醛,该文献并未提及该步骤的具体收率,同时路易斯酸作为催化剂对设备材质要求较高,产生的酸废液难于处理等问题,不是理想的工业化方案。
7.日本专利jph09192493(a)使用1,1,4,4-四甲氧基-2-丁烯和甲基-1-丙烯基醚在二草酸根合硼酸催化作用下反应生成四缩醛,其所述催化剂存在难以回收,催化剂可重复利用效果不佳等问题。
8.综上,目前合成四缩醛的方法存在产物四缩醛收率较低,反应原料烷基丙烯基醚在传统生产中存在容易变质的问题,造成该反应收率偏低,同时,副产物增多增大了生产成本和分离难度,使该工艺复杂性增大,路易斯酸作为催化剂对设备材质要求较高,产生的酸废液难于处理,催化剂难以回收,催化剂可重复利用效果不佳,不利于大规模工业化生产等问题。因此需要寻求一种新的四缩醛的合成方法解决上述技术问题。
技术实现要素:
9.本发明的目的是提供一种合成四缩醛的方法。该工艺的催化剂可循环使用,并且该工艺具有反应选择性高,副产物少,环境友好等优点。
10.为达到以上发明目的,本发明的技术方案如下:
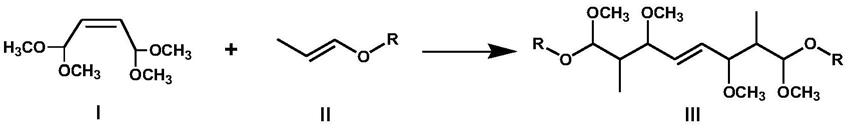
11.一种合成四缩醛的方法,包括以下步骤:式i的1,1,4,4-四甲氧基-2-丁烯(简称tmbu)和式ii的烷基丙烯基醚发生缩合反应生成式iii的四缩醛。
12.本发明式ii所述的烷基丙烯基醚,其中r可以为甲基、乙基、丙基、异丙基。
[0013][0014]
产物四缩醛的结构式为式iii,其中r为甲基、乙基、丙基、异丙基。
[0015][0016]
本发明所述的合成四缩醛的方法,反应是在催化剂条件下进行,所述催化剂以椰壳活性炭为载体,通过在所述椰壳活性炭上负载lewis酸、过渡金属硼化物、有机助剂制得,相对于反应体系,催化剂的质量用量为底物tmbu质量的0.01%~30%,优选0.05%~5%。
[0017]
作为一个优选的方案,本发明所述的合成四缩醛的方法,反应是在溶剂的存在下进行的,所述溶剂选自正己烷、正庚烷、石油醚(90-120)、甲苯、四氯化碳、氯仿、二氯甲烷中的一种或多种,优选石油醚(90-120)和/或甲苯。
[0018]
本发明所述的溶剂用量为tmbu质量的1~20倍,优选3~8倍。
[0019]
本发明所述的合成四缩醛的方法,反应温度为-50~100℃,优选温度为-30~50℃,反应时间包括烷基丙烯基醚滴加时间和滴加完保温时间,其中烷基丙烯基醚滴加时间为0.5~10h,优选2~5h;保温时间为0~4h,优选0.5~2h。
[0020]
本发明所述烷基丙烯基醚与tmbu的摩尔比为1.8~3.0,优选1.9~2.1。
[0021]
作为一个优选的方案,采用一种原料与催化剂、溶剂铺底,另一种原料通过滴加的方式加入反应体系,优选原料tmbu与催化剂、溶剂铺底,烷基丙烯基醚以滴加的方式加入反应体系。
[0022]
本发明所述的lewis酸选自过渡金属卤化物、第iiia族~第va族金属卤化物的一种或多种。合适的例子包括但不限于卤化铍、卤化硼、卤化钛、卤化钒、卤化亚铁、卤化铁、卤化钴、卤化镍、卤化亚铜、卤化铜、卤化锌、卤化银、卤化铝等中的一种或多种,优选卤化铍、卤化硼、卤化亚铁、卤化铁、卤化钴、卤化镍、卤化铜、卤化锌、卤化铝等中的一种或多种,更优化氯化铁、溴化铁、氯化锌、溴化锌、氯化铝等中的一种或多种。
[0023]
本发明所述的过渡金属硼化物选自feb、cob、nib、rub、rhb、pdb中的一种或多种。过渡金属硼化物的加入,可以提高催化剂的机械强度,使催化剂的寿命延长,循环套用次数增加。
[0024]
本发明所述的有机助剂选自4-羟基异佛尔酮、2-羟基-3,5,5-三甲基-2-环己烯-1-酮中的一种或多种。
[0025]
通过实验验证,催化剂中引入有机助剂可以降低反应产物iii与式ii的烷基丙烯基醚反应生成式iv的杂质,从而保证主产物式iii的选择性,推测其机理主要来自有机助剂和过渡金属硼化物共同作用下使烷基丙烯基醚与tmbu的反应更易发生,可以在较短的时间内将烷基丙烯基醚转化成式iii的主产物。
[0026]
[0027]
同时,对比不加有机助剂的反应,我们注意到加入有机助剂的反应在反应结束后检测不到过氧化物,众所周知,在工业化装置中过氧化物的存在较大的安全问题,因此有机助剂的加入最大限度的保证了工业化生产的安全。
[0028]
本发明所述催化剂的制备方法,包括以下步骤:
[0029]
将100份椰壳活性炭、50~150份lewis酸、5~60份过渡金属硼化物、2~20份有机助剂加入溶剂中,并于0~70℃条件下浸泡2~48h,将所得固体在80~150℃下烘干2~10h。
[0030]
优选的,本发明所述椰壳活性炭在用于制备催化剂之前,使用去离子水浸泡搅拌清洗,然后在100~150℃下活化1~5h。冷却至室温,将椰壳活性炭滤出备用。所述去离子水与椰壳活性炭的质量比为1~5:1,优选2~4:1。
[0031]
本发明所述制备催化剂的方法中,所述溶剂为本领域公知的溶剂,优选甲醇、乙醇、正丙醇、异丙醇等脂肪族醇类,更优选甲醇。
[0032]
本发明所述“份”为“质量份”。
具体实施方式
[0033]
下面的实施例将对本发明所提供的方法予以进一步的说明,但本发明不限于所列出的实施例,还应包括在本发明所要求的权利范围内其它任何公知的改变。
[0034]
气相色谱分析条件:安捷伦气相色谱,色谱柱db-5进行在线测定,二阶程序升温,初始温度50℃,保持1分钟后以5℃/min的速率升至80℃;再以10℃/min的速率升至250℃。载气高纯n2,分流比100:1。进样温度250℃,检测器为fid,检测器温度250℃。
[0035]
过氧化值检测方法:
[0036]
步骤:
[0037]
(1)准确称取5.0g~10.0g(精确至0.1mg)样品于250ml的碘量瓶中。
[0038]
(2)用量筒量分别量取10ml正己烷、20ml冰醋酸、60ml异丙醇、20ml甲醇、20ml碘化钾水溶液(20g/l)加入到碘量瓶中,混合均匀,水封后避光静置20分钟。
[0039]
(3)将碘量瓶中液体转移至200ml一次性塑料杯中,用30ml异丙醇分三次冲洗碘量瓶,并将清洗液一并转移至塑料杯中。
[0040]
(4)在电位滴定仪上,以0.1mol/l硫代硫酸钠标准溶液(0.1mol/l)滴定至ep1。
[0041]
(5)不加样品,重复3.2~3.4步骤,做空白试验。
[0042]
计算及结果表述
[0043]
分析结果按下式计算(以过氧化氢百分含量计):
[0044]
w=(v-v0)
×c×
17.01
÷m÷
1000
×
100%
[0045]
其中
[0046]
w—样品中的过氧化物百分含量,%;
[0047]
v—滴定样品时消耗硫代硫酸钠标准溶液的体积,ml;
[0048]v0
—滴定空白时消耗硫代硫酸钠标准溶液的体积,ml;
[0049]
c—硫代硫酸钠标准溶液的浓度
[0050]
m—试样质量,g;
[0051]
17.01—1/2过氧化氢的摩尔质量。
[0052]
5.2允许误差
[0053]
取2次平行测定结果的算术平均值为测定结果,两次绝对结果之差《0.0020%。
[0054]
实施例1cat-1制备
[0055]
将300g椰壳活性炭用900g的去离子水浸泡搅拌清洗,随后在110℃下活化5h。冷却至室温,将活化后椰壳活性炭滤出备用。
[0056]
称取100g活化后椰壳活性炭、100g溴化铁、30g feb、15g 2-羟基-3,5,5-三甲基-2-环己烯-1-酮,加入1.0kg无水甲醇中,并于30℃条件下浸泡20h,加压过滤。将所得固体分散均匀后在120℃下烘干4h,冷却至室温备用,所得催化剂记为cat-1。
[0057]
实施例2-4cat-2-4制备
[0058]
cat-2-4制备工艺步骤参考实施例1,相关物料及参数如下表1。
[0059]
表1 cat-2-4相关物料及参数
[0060][0061]
实施例5合成化合物iii
[0062]
依次在2000ml四口烧瓶中称量88.1g tmbu、440.4g甲苯与1.76g cat-1,将四口烧瓶放置在油浴中,开启机械搅拌,控制反应液内温恒定在25℃,开始滴加甲基丙烯基醚,甲基丙烯基醚总质量75.7g,滴加时间2.5h,滴加完继续25℃保温1h,取样进行气相分析,得到原料tmbu转化率99.2%,化合物iii(r=ch3)选择性98.1%,反应液中过氧化值未检出。
[0063]
实施例6-8合成化合物iii
[0064]
合成工艺步骤参考实施例5,相关物料及参数如下表2。
[0065]
表2实施例6-8相关物料及参数
[0066]
[0067][0068]
备注:烷基丙烯基醚中的r=ch3时,式iii的r=ch3;烷基丙烯基醚中的r=ch2ch3时,式iii的r=ch2ch3。
[0069]
实施例9-18
[0070]
实施例9-18按照实施例5的工艺进行重复反应,实施例9的催化剂使用实施例5滤出的催化剂,实施例10的催化剂使用实施例9滤出的催化剂,以此类推,催化剂循环使用,考察催化剂循环使用效果,其结果如下表3所示:
[0071]
表3催化剂循环套用结果
[0072]
实施例tmbu转化率/%化合物iii选择性/%过氧化值/%999.298.1未检出1099.198.3未检出1199.498.2未检出1299.198.0未检出1399.398.0未检出1499.398.4未检出1599.098.1未检出1699.198.3未检出1799.398.2未检出1899.298.3未检出
[0073]
由上表,cat-1重复使用10次后,原料tmbu转化率和产物化合物iii选择性无明显降低,反应效果与初始反应(实施例5)相比可以维持。
[0074]
对比例1(不使用有机助剂)
[0075]
将300g椰壳活性炭用900g的去离子水浸泡搅拌清洗,随后在110℃下活化5h。冷却至室温,将活化后椰壳活性炭滤出备用。
[0076]
称取100g活化后椰壳活性炭、100g溴化铁、30g feb,加入1.0kg无水甲醇中,并于30℃条件下浸泡20h,加压过滤。将所得固体分散均匀后在120℃下烘干4h,冷却至室温备用,所得催化剂记为cat-5。
[0077]
依次在2000ml四口烧瓶中称量88.1g tmbu、440.4g甲苯与1.76g cat-5,将四口烧瓶放置在油浴中,开启机械搅拌,控制反应液内温恒定在25℃,开始滴加甲基丙烯基醚,甲基丙烯基醚总质量75.7g,滴加时间2.5h,滴加完继续25℃保温1h,取样进行气相分析,得到原料tmbu转化率89.4%,化合物iii(r=ch3)选择性87.9%,反应液中过氧化值2650mgh2o2/kg。
[0078]
对比例2(不使用过渡金属硼化物)
[0079]
将300g椰壳活性炭用900g的去离子水浸泡搅拌清洗,随后在110℃下活化5h。冷却至室温,将活化后椰壳活性炭滤出备用。
[0080]
称取100g活化后椰壳活性炭、100g溴化铁、15g 2-羟基-3,5,5-三甲基-2-环己烯-1-酮,加入1.0kg无水甲醇中,并于30℃条件下浸泡20h,加压过滤。将所得固体分散均匀后在120℃下烘干4h,冷却至室温备用,所得催化剂记为cat-6。
[0081]
依次在2000ml四口烧瓶中称量88.1g tmbu、440.4g甲苯与1.76g cat-6,将四口烧瓶放置在油浴中,开启机械搅拌,控制反应液内温恒定在25℃,开始滴加甲基丙烯基醚,甲基丙烯基醚总质量75.7g,滴加时间2.5h,滴加完继续25℃保温1h,取样进行气相分析,得到原料tmbu转化率93.1%,化合物iii(r=ch3)选择性93.7%,反应液中过氧化值未检出。
[0082]
对比例3、4
[0083]
对上述对比例2滤出的催化剂cat-6重复使用,反应工艺步骤与对比例2相同,其反应结果如下表4:
[0084]
表4 cat-6套用结果
[0085]
对比例tmbu转化率/%化合物iii选择性/%过氧化值/%391.592.4未检出479.490.1未检出
[0086]
对比例5(不使用过渡金属硼化物和有机助剂)
[0087]
将300g椰壳活性炭用900g的去离子水浸泡搅拌清洗,随后在110℃下活化5h。冷却至室温,将活化后椰壳活性炭滤出备用。
[0088]
称取100g活化后椰壳活性炭、100g溴化铁,加入1.0kg无水甲醇中,并于30℃条件下浸泡20h,加压过滤。将所得固体分散均匀后在120℃下烘干4h,冷却至室温备用,所得催化剂记为cat-7。
[0089]
依次在2000ml四口烧瓶中称量88.1g tmbu、440.4g甲苯与1.76g cat-7,将四口烧瓶放置在油浴中,开启机械搅拌,控制反应液内温恒定在25℃,开始滴加甲基丙烯基醚,甲基丙烯基醚总质量75.7g,滴加时间2.5h,滴加完继续25℃保温1h,取样进行气相分析,得到原料tmbu转化率77.6%,化合物iii(r=ch3)选择性85.1%,反应液中过氧化值3349mgh2o2/kg。