一种制备5-溴-2-氯-4
’‑
乙氧基二苯甲烷的合成方法
技术领域
1.本发明涉及一种制备5-溴-2-氯-4
’‑
乙氧基二苯甲烷的合成方法,属于医药中间体制备技术领域。
背景技术:
2.据申请人所知,达格列净是由百时美施贵宝和阿斯利康公司联合开发的一种新型的抗糖尿病药物,于2012年11月12日被欧洲药品管理局(ema)批准上市,是第1个获准上市用于治疗2型糖尿病的sglt2抑制剂,可作为糖尿病药物治疗中的重要选择,适用在有2型糖尿病成人中作为辅助饮食和运动改善血糖控制。美国食品药品管理局(fda)于2014年1月8日宣布,批准将达格列净用于2型糖尿病的治疗。达格列净通过抑制钠-葡萄糖转运蛋白2(sglt2)——肾内的一种使葡萄糖被重新吸收到血液中的蛋白质——而发挥作用;这使得多余的葡萄糖通过尿液被排出体外,从而在不增加胰岛素分泌的情况下改善血糖控制。而5-溴-2-氯-4'-乙氧基二苯甲烷是制备达格列净的重要中间体,因此该中间体市场表现日益走强,达到供不应求的状况。
3.关于5-溴-2-氯-4'-乙氧基二苯甲烷的制备方法,文献中有很多报道。例如:申请号cn201410664465.7、申请公布号cn104478670a的发明专利申请,以邻甲苯胺经溴化、重氮化、桑德迈尔、卤代、傅克烷基化合成,其中卤代使用的偶氮二异丁腈为剧毒化合物,对人及环境伤害重大。又如:申请号cn201710566635.1、申请公布号cn107311847a的发明专利申请,以4-羟基苯甲醛与溴乙烷作为原料,经取代、缩合、重氮化、桑德迈尔、还原合成,虽然原料为常见原料,但所用辅料及溶剂较多且繁琐,收率低。此外还有技术方案以5-溴-2-氯苯甲酸为起始原料,经酰氯化,傅克酰基化再还原所得,其中原料成本价格高,收率不高,不利于生产。
技术实现要素:
4.本发明的主要目的是:克服现有技术存在的问题,提供一种制备5-溴-2-氯-4
’‑
乙氧基二苯甲烷的合成方法,以邻氯苯甲酸为起始原料,路线精简,所用原料、辅料均为常见化合物,采用低毒性溶剂且循环回收利用,不会产生含磷废水,安全性高且对环境友好,所得产品纯度高。
5.本发明解决其技术问题的技术方案如下:一种制备5-溴-2-氯-4
’‑
乙氧基二苯甲烷的合成方法,其特征是,包括以下步骤:第一步、取邻氯苯甲酸,先加入溶剂和催化剂,再滴加酰氯化试剂,经反应并回收溶剂后获得邻氯苯甲酰氯;其中,邻氯苯甲酸与溶剂的重量比为1:4.0-6.0,邻氯苯甲酸与催化剂的重量比为1:0.003-0.006,邻氯苯甲酸与酰氯化试剂的摩尔比为1:1.05-1.15;第二步、取邻氯苯甲酰氯,加入溶剂并以惰性气体保护,加入三氯化铝,滴加碘苯后反应,反应后淬灭、萃取、洗涤并回收溶剂后获得2-氯-4'-碘二苯甲酮;其中,邻氯苯甲酸氯与溶剂的重量比为1:1.9-4.0,邻氯苯甲酸氯与三氯化铝的摩尔比为1:1.05-1.15,邻氯
苯甲酸氯与碘苯的摩尔比为1:1.00-1.05;第三步、取2-氯-4'-碘二苯甲酮,加入酸和催化剂,加入溴化剂后反应,反应后淬灭并获得粗品,将粗品以石油醚精制,获得5-溴-2-氯-4'-碘二苯甲酮;其中,2-氯-4'-碘二苯甲酮与酸的重量比为1:3.0-5.0,2-氯-4'-碘二苯甲酮与催化剂的重量比为1:0.0003-0.0005,2-氯-4'-碘二苯甲酮与溴化剂的摩尔比为1:1.00-1.05;粗品与石油醚的重量比为1:0.4-0.6;第四步、取5-溴-2-氯-4'-碘二苯甲酮,加入溶剂,加入还原剂和三氯化铝后反应,反应后回收溶剂、萃取、洗涤,并将所得产物以无水乙醇精制,获得5-溴-2-氯-4'-碘二苯甲烷;其中,5-溴-2-氯-4'-碘二苯甲酮与溶剂的重量比为1:2.9-5.0,5-溴-2-氯-4'-碘二苯甲酮与还原剂的摩尔比为1:1.3-1.8,5-溴-2-氯-4'-碘二苯甲酮与三氯化铝的摩尔比为1:1.7-2.2;所得产物与无水乙醇的重量比为1:0.7-0.9;第五步、取5-溴-2-氯-4'-碘二苯甲烷,加入溶剂,滴加乙醇钠后反应,反应后回收溶剂并析出固体,加水后抽滤并烘干,获得5-溴-2-氯-4'-乙氧基二苯甲烷;其中,5-溴-2-氯-4'-碘二苯甲烷与溶剂的重量比为1:2.0-4.0,5-溴-2-氯-4'-碘二苯甲烷与乙醇钠的摩尔比为1:1.00-1.05。
6.该方法中,先将邻氯苯甲酸氯化制成邻氯苯甲酰氯,再傅克酰基化制成2-氯-4'-碘二苯甲酮,进一步溴化制成5-溴-2-氯-4'-碘二苯甲酮,后经还原制成5-溴-2-氯-4'-碘二苯甲烷,最终经取代反应制成5-溴-2-氯-4'-乙氧基二苯甲烷,所得产品纯度在99.3%以上。该方法路线精简,所用原料、辅料均为常见化合物,采用低毒性溶剂且能循环回收利用(即套用),不会产生含磷废水,安全性高且对环境友好,成本低,对设备要求不高,适合工业化生产。
7.本发明进一步完善的技术方案如下:优选地,第一步中,加入溶剂和催化剂后搅拌均匀,在0℃-10℃下滴加酰氯化试剂;在20℃-30℃下反应2-4小时;反应结束后在40℃-70℃下减压蒸馏回收溶剂;所述溶剂为二氯乙烷、甲苯或二氯甲烷;所述催化剂为吡啶或dmf;所述酰氯化试剂为氯化亚砜或草酰氯。
8.更优选地,第一步中,反应终点为hplc检测邻氯苯甲酸《0.2%;所述溶剂为二氯甲烷;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.3%;所述催化剂为吡啶;所述酰氯化试剂为草酰氯;所述邻氯苯甲酰氯呈油状物。
9.采用以上优选方案后,可进一步优化第一步的具体参数。
10.优选地,第二步中,加入溶剂并以惰性气体保护后,在-5℃-0℃下分批加入三氯化铝,滴加碘苯后在0℃-5℃下反应2-4小时;反应结束时倒入冰水中淬灭同时控制温度为10℃-30℃;萃取、并以质量分数3.6-6.0%盐酸洗涤后,萃取合并有机层并在40℃-70℃下减压蒸馏回收溶剂;所述溶剂为二氯乙烷、甲苯或二氯甲烷。
11.更优选地,第二步中,所述惰性气体为氮气;反应终点为hplc检测邻氯苯甲酸氯《0.3%;所述溶剂为二氯甲烷;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.3%;所述2-氯-4'-碘二苯甲酮呈油状物。
12.采用以上优选方案后,可进一步优化第二步的具体技术特征。
13.优选地,第三步中,加入酸和催化剂后,在0℃-10℃下分批加入溴化剂;在10℃-25
℃下反应4-6小时;反应结束时倒入冰水中淬灭并控制温度为10℃-30℃;降温至0℃-10℃,抽滤获得粗品;以石油醚精制时,先将粗品溶于石油醚,再升温至40℃-60℃并保持至少1小时,然后逐渐降温至-5℃-0℃以析出晶体,抽滤并将固体在40℃-60℃下烘干,精制结束;所述酸为浓盐酸或浓硫酸;所述催化剂为碘或bpo;所述溴化剂为nbs或dmbs。
14.更优选地,第三步中,反应终点为hplc检测2-氯-4'-碘二苯甲酮《0.5%;所述酸为浓硫酸;所述催化剂为碘;所述溴化剂为nbs;所述5-溴-2-氯-4'-碘二苯甲酮呈固体。
15.采用以上优选方案后,可进一步优化第三步的具体参数。其中,bpo为过氧化苯甲酰,nbs为n-溴代琥珀酰亚胺,dmbs为溴化二甲基溴化硫。
16.优选地,第四步中,加入溶剂后,在-5℃-0℃下加入还原剂和三氯化铝,在-5℃-0℃下反应1-3小时,之后在60℃-80℃回流反应10-16小时;反应后在40℃-60℃下减压蒸馏回收溶剂;加入温度《20℃的水降温,以二氯甲烷萃取,并以质量分数3.6-6.0%盐酸洗涤,合并有机层并在40℃-60℃下减压蒸馏回收二氯甲烷,获得呈油状物的产物;以无水乙醇精制时,先将所得产物溶于无水乙醇,再升温至40℃-60℃并保持至少1小时,然后逐渐降温至-5℃-0℃以析出晶体,抽滤并将固体在40℃-60℃下烘干,精制结束;所述溶剂为异丙醇、甲苯或四氢呋喃;所述还原剂为异丙醇铝或硼氢化钠。
17.更优选地,第四步中,反应终点为hplc检测5-溴-2-氯-4'-碘二苯甲酮《0.3%;所述溶剂为四氢呋喃;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.3%;所述还原剂为硼氢化钠;所述5-溴-2-氯-4'-碘二苯甲烷呈固体。
18.采用以上优选方案后,可进一步优化第四步的具体参数。
19.优选地,第五步中,加入溶剂后,在0℃-10℃下滴加乙醇钠,在40℃-80℃回流反应3-5小时,在40℃-60℃下减压蒸馏回收溶剂,加水降温至-5℃-0℃后抽滤,并在30℃-40℃下烘干;所述溶剂为甲醇或乙醇;所述乙醇钠采用浓度为重量比27-30%的乙醇钠溶液。
20.更优选地,第五步中,反应终点为hplc检测5-溴-2-氯-4'-碘二苯甲烷《0.3%;所述溶剂为乙醇;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.5%;所述5-溴-2-氯-4'-乙氧基二苯甲烷呈固体。
21.采用以上优选方案后,可进一步优化第五步的具体参数。
22.与现有技术相比,本发明采用以邻氯苯甲酸为起始原料制备5-溴-2-氯-4'-乙氧基二苯甲烷的精简路线,所得产品纯度高(99.3%以上)利于销售;所用原料、辅料均为常见化合物,易于获得;采用低毒性溶剂且能循环回收利用(即套用),不会产生含磷废水,安全性高且对环境友好,成本低,对设备要求不高,适合工业化生产。
附图说明
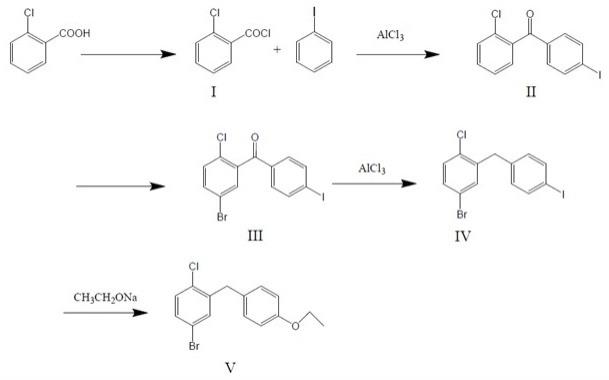
23.图1为本发明具体实施时的工艺路线图。
24.图2为本发明实施例6中5-溴-2-氯-4
’‑
乙氧基二苯甲烷的1h nmr图。
具体实施方式
25.如图1所示,具体实施时,本发明制备5-溴-2-氯-4
’‑
乙氧基二苯甲烷的合成方法包括:第一步、取邻氯苯甲酸,先加入溶剂和催化剂,再滴加酰氯化试剂,经反应并回收
溶剂后获得邻氯苯甲酰氯(图1的化合物i);其中,邻氯苯甲酸与溶剂的重量比为1:4.0-6.0,邻氯苯甲酸与催化剂的重量比为1:0.003-0.006,邻氯苯甲酸与酰氯化试剂的摩尔比为1:1.05-1.15。
26.具体而言,加入溶剂和催化剂后搅拌均匀,在0℃-10℃下滴加酰氯化试剂;在20℃-30℃下反应2-4小时;反应结束后在40℃-70℃下减压蒸馏回收溶剂;溶剂为二氯乙烷、甲苯或二氯甲烷(优选二氯甲烷);催化剂为吡啶或dmf(优选吡啶);酰氯化试剂为氯化亚砜或草酰氯(优选草酰氯)。反应终点为hplc检测邻氯苯甲酸《0.2%;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.3%;所得邻氯苯甲酰氯呈油状物。
27.第二步、取邻氯苯甲酰氯,加入溶剂并以惰性气体保护,加入三氯化铝,滴加碘苯后反应,反应后淬灭、萃取、洗涤并回收溶剂后获得2-氯-4'-碘二苯甲酮(图1的化合物ii);其中,邻氯苯甲酸氯与溶剂的重量比为1:1.9-4.0,邻氯苯甲酸氯与三氯化铝的摩尔比为1:1.05-1.15,邻氯苯甲酸氯与碘苯的摩尔比为1:1.00-1.05。
28.具体而言,加入溶剂并以惰性气体(优选氮气)保护后,在-5℃-0℃下分批加入三氯化铝,滴加碘苯后在0℃-5℃下反应2-4小时;反应结束时在10℃-30℃下淬灭;萃取、并以质量分数3.6-6.0%盐酸洗涤后在40℃-70℃下减压蒸馏回收溶剂;溶剂为二氯乙烷、甲苯或二氯甲烷(优选二氯甲烷)。反应终点为hplc检测邻氯苯甲酸氯《0.3%;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.3%;所得2-氯-4'-碘二苯甲酮呈油状物。
29.第三步、取2-氯-4'-碘二苯甲酮,加入酸和催化剂,加入溴化剂后反应,反应后淬灭并获得粗品,将粗品以石油醚精制,获得5-溴-2-氯-4'-碘二苯甲酮(图1的化合物iii);其中,2-氯-4'-碘二苯甲酮与酸的重量比为1:3.0-5.0,2-氯-4'-碘二苯甲酮与催化剂的重量比为1:0.0003-0.0005,2-氯-4'-碘二苯甲酮与溴化剂的摩尔比为1:1.00-1.05;粗品与石油醚的重量比为1:0.4-0.6。
30.具体而言,加入酸和催化剂后,在0℃-10℃下分批加入溴化剂;在10℃-25℃下反应4-6小时;反应结束时在0℃-10℃下淬灭,然后抽滤获得粗品;以石油醚精制时,先将粗品溶于石油醚,再升温至40℃-60℃并保持至少1小时,然后逐渐降温至-5℃-0℃以析出晶体,抽滤并将固体在40℃-60℃下烘干,精制结束;酸为浓盐酸或浓硫酸(优选浓硫酸);催化剂为碘或bpo(优选碘);溴化剂为nbs或dmbs(优选nbs);反应终点为hplc检测2-氯-4'-碘二苯甲酮《0.5%;所得5-溴-2-氯-4'-碘二苯甲酮呈固体。
31.第四步、取5-溴-2-氯-4'-碘二苯甲酮,加入溶剂,加入还原剂和三氯化铝后反应,反应后回收溶剂、萃取、洗涤,并将所得产物以无水乙醇精制,获得5-溴-2-氯-4'-碘二苯甲烷(图1的化合物iv);其中,5-溴-2-氯-4'-碘二苯甲酮与溶剂的重量比为1:2.9-5.0,5-溴-2-氯-4'-碘二苯甲酮与还原剂的摩尔比为1:1.3-1.8,5-溴-2-氯-4'-碘二苯甲酮与三氯化铝的摩尔比为1:1.7-2.2;所得产物与无水乙醇的重量比为1:0.7-0.9。
32.具体而言,加入溶剂后,在-5℃-0℃下加入还原剂和三氯化铝,在-5℃-0℃下反应1-3小时,之后在60℃-80℃回流反应10-16小时;反应后在40℃-60℃下减压蒸馏回收溶剂;加入温度《20℃的水降温,以二氯甲烷萃取,并以质量分数3.6-6.0%盐酸洗涤,合并有机层并在40℃-60℃下减压蒸馏回收二氯甲烷,获得呈油状物的产物;以无水乙醇精制时,先将所得产物溶于无水乙醇,再升温至40℃-60℃并保持至少1小时,然后逐渐降温至-5℃-0℃以析出晶体,抽滤并将固体在40℃-60℃下烘干,精制结束;溶剂为异丙醇、甲苯或四氢呋喃
(优选四氢呋喃);还原剂为异丙醇铝或硼氢化钠(优选硼氢化钠)。反应终点为hplc检测5-溴-2-氯-4'-碘二苯甲酮《0.3%;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.3%;所得5-溴-2-氯-4'-碘二苯甲烷呈固体。
33.第五步、取5-溴-2-氯-4'-碘二苯甲烷,加入溶剂,滴加乙醇钠后反应,反应后回收溶剂并析出固体,加水后抽滤并烘干,获得5-溴-2-氯-4'-乙氧基二苯甲烷(图1的化合物v);其中,5-溴-2-氯-4'-碘二苯甲烷与溶剂的重量比为1:2.0-4.0,5-溴-2-氯-4'-碘二苯甲烷与乙醇钠的摩尔比为1:1.00-1.05。
34.具体而言,加入溶剂后,在0℃-10℃下滴加乙醇钠,在40℃-80℃回流反应3-5小时,在40℃-60℃下减压蒸馏回收溶剂,加水降温至-5℃-0℃后抽滤,并在30℃-40℃下烘干;溶剂为甲醇或乙醇(优选乙醇);乙醇钠采用浓度为重量比27-30%的乙醇钠溶液。反应终点为hplc检测5-溴-2-氯-4'-碘二苯甲烷《0.3%;回收后溶剂套用的标准为:gc检测纯度99.8%以上,kf检测水分低于0.5%;所得5-溴-2-氯-4'-乙氧基二苯甲烷呈固体。
35.下面结合实施例对本发明作进一步详细描述。但是本发明不限于所给出的例子。
36.实施例1本实施例为制备邻氯苯甲酰氯(图1的化合物i)。
37.本实施例基本过程为具体实施时本发明技术方案的第一步。
38.本实施例的一些具体参数如下:在2000ml四口反应瓶中加入邻氯苯甲酸156.0g(1.0mol)和二氯甲烷800g,dmf0.8g,开启搅拌,降温至10℃,滴加草酰氯139.7g(1.1mol),滴完室温(20℃-30℃)反应3小时(检测原料hplc《0.2%为止),蒸去溶剂,得到油状物,主要为邻氯苯甲酰氯,以及很少量的草酰氯;该油状物经hplc检测纯度为99.0%,该纯度按照hplc面积归一化法计算,计算时扣除了溶剂峰,同时由于在hplc中草酰氯是不出峰的,所以hplc纯度检测结果较高。该油状物中含有邻氯苯甲酰氯172.4g,收率为98.5%(产品理论重量为175.0g)。
39.实施例2本实施例为制备2-氯-4'-碘二苯甲酮(图1的化合物ii)。
40.本实施例基本过程为具体实施时本发明技术方案的第二步。
41.本实施例的一些具体参数如下:在1000ml四口反应瓶中加入实施例1所得油状物(含邻氯苯甲酰氯172.4g(0.985mol))和二氯甲烷350.0g,氮气保护,降温至0℃,分批加入三氯化铝144.6g(1.08mol),滴加碘苯205.0g(1.00mol),0℃-5℃反应2小时(检测原料hplc《0.3%为止),结束后倒入冰水中(控制加入温度《20℃),由于三氯化铝倒入水中会剧烈放热,而温度过高会导致杂质含量过高,因此要控制温度在10℃-30℃之间。萃取、并以质量分数3.6%盐酸洗涤,萃取合并有机层蒸去溶剂,得到油状物,主要为2-氯-4'-碘二苯甲酮;该油状物经hplc检测纯度为97.0%,该纯度按照hplc面积归一化法计算,计算时扣除了溶剂峰。该油状物含有2-氯-4'-碘二苯甲酮315.4g,收率为93.5%(产品理论重量为337.4g)。
42.实施例3本实施例为制备5-溴-2-氯-4'-碘二苯甲酮(图1的化合物iii)。
43.本实施例基本过程为具体实施时本发明技术方案的第三步。
44.本实施例的一些具体参数如下:
在2000ml四口反应瓶中加入实施例2所得油状物(含2-氯-4'-碘二苯甲酮315.4g(0.92mol))332.8g(0.97mol)和硫酸1000.0g,碘0.1g,搅拌,降温至10℃分批加入nbs 171.0g(0.96mol),加完后在10℃-15℃反应4小时(检测原料hplc《0.5%为止),结束后缓慢倒入冰水中淬灭,温度控制在10℃-30℃,由于硫酸倒入水中会剧烈放热,而温度过高会导致杂质含量过高,因此要控制温度在10℃-30℃之间。之后继续降温至0℃-10℃,抽滤得粗品;粗品加石油醚(0.5倍粗品重量),升温至60℃1小时,慢慢降温析出晶体、慢慢降至-5℃-0℃,抽滤、烘干,得到固体,主要为5-溴-2-氯-4'-碘二苯甲酮;该固体经hplc检测纯度为99.3%,该纯度按照hplc面积归一化法计算,计算时扣除了溶剂峰。该固体含有5-溴-2-氯-4'-碘二苯甲酮345.1g,收率为89.0%(产品理论重量为387.8g)。
45.实施例4本实施例为制备5-溴-2-氯-4'-碘二苯甲烷(图1的化合物iv)。
46.本实施例基本过程为具体实施时本发明技术方案的第四步。
47.本实施例的一些具体参数如下:在2000ml四口反应瓶中加入5-溴-2-氯-4'-碘二苯甲酮210.8g(0.5mol),四氢呋喃650.0g,降温至-5℃加入硼氢化钠27.8g(0.75mol),分批加入三氯化铝133.5g(1.0mol)加完后-5-0℃反应1小时,升温至70℃反应14小时(检测原料hplc《0.3%为止),蒸去溶剂,缓慢加冷水(控制加入温度《20℃),二氯甲烷萃取,质量分数3.6-6.0%盐酸洗涤,合并有机层,蒸去溶剂,加入无水乙醇(0.8倍油状产物重量),升温至60℃1小时,慢慢降温析出晶体、慢慢降至-5-0℃抽滤、烘干,得到固体,主要为5-溴-2-氯-4'-碘二苯甲烷;该固体经hplc检测纯度为99.3%,该纯度按照hplc面积归一化法计算,计算时扣除了溶剂峰。该固体含有5-溴-2-氯-4'-碘二苯甲烷179.5g,收率为88.1%(产品理论重量为203.8g)。
48.实施例5本实施例为以回收有机溶剂制备5-溴-2-氯-4'-碘二苯甲烷(图1的化合物iv)。
49.本实施例基本过程为具体实施时本发明技术方案的第四步。
50.本实施例的一些具体参数如下:在2000ml四口反应瓶中加入5-溴-2-氯-4'-碘二苯甲酮168.6g(0.4mol),回收的四氢呋喃500.0g,降温至-5℃加入硼氢化钠22.2g(0.6mol),分批加入三氯化铝106.8g(0.8mol)加完后-5-0℃反应1小时,升温至70℃反应14小时(检测原料hplc《0.3%为止),蒸去溶剂,缓慢加冰水(控制加入温度《20℃),用回收的二氯甲烷萃取,质量分数3.6-6.0%盐酸洗涤,合并有机层,蒸去溶剂,加入无水乙醇(0.8倍油状产物重量)升温至60℃1小时,慢慢降温析出晶体、慢慢降至-5-0℃抽滤、烘干,得到固体,主要为5-溴-2-氯-4'-碘二苯甲烷;该固体经hplc检测纯度为99.2%,该纯度按照hplc面积归一化法计算,计算时扣除了溶剂峰。该固体含有5-溴-2-氯-4'-碘二苯甲烷144.3g,收率为88.5%(产品理论重量为163.0g)。
51.实施例6本实施例为制备5-溴-2-氯-4'-乙氧基二苯甲烷(图1的化合物v)。
52.本实施例基本过程为具体实施时本发明技术方案的第五步。
53.本实施例的一些具体参数如下:在1000ml四口反应瓶中加入5-溴-2-氯-4'-碘二苯甲烷163.0g(0.4mol),无水乙
醇350.0g,0℃-10℃下滴加30%乙醇钠93.0g,升温至40℃-80℃回流反应(检测原料hplc《0.3%为止),结束后减压蒸去乙醇至析出固体,加水再慢慢降至-5-0℃,抽滤烘干,得到固体,主要为5-溴-2-氯-4'-乙氧基二苯甲烷;该固体经hplc检测纯度为99.4%,该纯度按照hplc面积归一化法计算,计算时扣除了溶剂峰。该固体含有5-溴-2-氯-4'-乙氧基二苯甲烷125.1g,收率为96.1%(产品理论重量为130.2g)。
54.本实施例所得5-溴-2-氯-4'-乙氧基二苯甲烷的1h nmr 400mhz cdcl3图谱如图2所示:1hnmr(400mhz,cdcl3)δ=7.24-7.32(m,3h,arh),7.12(d,2h,arh),6.86(d,2h,arh),4.02-4.06(m,4h,ch2),1.44(t,3h,ch3)。
55.除上述实施例外,本发明还可以有其他实施方式。凡采用等同替换或等效变换形成的技术方案,均落在本发明要求的保护范围。