糖基转移酶osugt91c1突变体及其应用
技术领域
1.本发明属于酶学领域,具体涉及一种糖基转移酶osugt91c1突变体及其应用。
背景技术:
2.近年来高热量糖的危害逐步得到重视,但由于甜味可刺激产生内源性阿片、多巴胺等奖励性神经递质,使人难于戒除对甜味的依赖。几种量产的人造甜味剂虽可满足热量低的需求,但口感、安全性和心理接受度仍存在争议,难以有效替代传统高热量甜味剂。原产于南美的甜叶菊在当地作为天然甜味来源已有超过百年的历史。2006年世界卫生组织(who)和联合国粮农组织(fao)联合食品添加剂专用委员会(jecfa)通过了甜菊糖健康无害的安全评价。2008年美国fda正式批准允许甜菊糖在食品中使用。2011年甜菊糖取得了欧洲的食品安全局安全性检验证书。随着消费者对低糖、低热量的食品和饮料的需求持续增长,甜菊糖已成为全球使用量增长最快的天然甜味剂之一。
3.甜菊糖是多种甜菊糖苷的总称。所有甜菊糖苷具有相似的化学结构模式,即在通用的甜菊醇(steviol,cas 471-80-7)核心骨架c13-羟基(为了简化,在附图中也用r1代表)和(或)c19-羧基(为了简化,在附图中也用r2代表)存在不同的葡萄糖基修饰(图1中a)。根据c13-羟基和c19-羧基的糖基组成和糖苷键的组合情况,可对甜菊糖苷分子进行命名,但目前市场上的甜菊糖产品以甜菊苷(stevioside,cas 57817-89-7占比57%,用st表示)和甜菊糖苷a(rebaudioside a,cas 58543-16-1,含量可达甜菊糖苷的32%,用reb a表示)为代表,二者存在口感不佳的劣势,影响了甜菊糖苷的市场接受度。研究发现甜菊糖苷d(rebaudioside d,cas 63279-13-0,reb d代表)和m(rebaudioside m,cas 1220616-44-3,reb m代表)甜度是蔗糖的200-300倍,口感非常接近于甜味标准品蔗糖,被认为是甜菊糖苷品质最好的组分,也是甜菊糖苷生产商研发的重点。百事可乐和可口可乐都获得了在饮料中使用甜菊糖苷d和m的专利。
4.然而,甜菊糖苷d和m在天然甜叶菊叶片中的含量低于1%,难于直接从天然种植的甜叶菊提取。由于含量低,从甜叶菊提取过程繁琐,成本高,限制了口感最好的甜菊糖苷d和m的市场化应用。研究表明甜叶菊自身缺少在甜菊糖苷的c19-羧基方向添加2号葡萄糖基的催化能力,不能有效合成reb d和reb m,导致了二者天然含量低的现象;同时reb d和reb m在c19-羧基方向的2号葡萄糖基,使二者同其他甜菊糖苷相比,口感得以显著提升的原因。因此,利用异源酶促转化的方法弥补甜叶菊催化在甜菊糖苷的c19-羧基方向添加2号葡萄糖基的能力不足,是规模化制备甜菊糖苷d和m的解决手段。一种水稻来源的糖基转移酶osugt91c1(也可称为eugt11),可催化在甜菊糖苷的c13-羟基和c19-羧基方向添加2号葡萄糖基(2号葡萄糖基是与1号葡萄糖基形成β(1-2)糖苷键的葡萄糖基,1号葡萄糖基特指直接与c13-羟基或c19-羧基形成β-糖苷键的葡萄糖基)(图1中b,c,d)。利用糖基转移酶osugt91c1在甜菊糖苷的c13-羟基和c19-羧基两个方向都可添加2号葡萄糖基的能力,弥补甜叶菊自身在甜菊糖苷的c19-羧基方向添加2号葡萄糖基能力的不足,是能否规模化生产甜菊糖苷d和m的决定因素。通过包含糖基转移酶osugt91c1的一系列酶促反应,在生物体内
(不一定是甜叶菊)或生物体外利用前体物质进行甜菊糖苷的全合成转化,或通过酶促反应对甜叶菊天然提取物,特别是在自然含量较为丰富的甜菊苷(st)、甜菊糖苷a(reb a)或其他可利用的甜菊糖苷分子的特定位置添加葡萄糖基,转化生成甜菊糖苷d和m(reb d和reb m)。
5.由于在甜菊糖苷的c19-羧基方向添加2号葡萄糖基是生成reb d和reb m的关键,发明人希望能进一步提高糖基转移酶osugt91c1在甜菊糖苷c19-羧基方向催化添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的能力,用于更加高效地转化生成甜菊糖苷d和m。同时,发明人发现糖基转移酶osugt91c1在催化甜菊糖苷d、m酶促转化过程中的特异性不好,除了添加2号葡萄糖基外(这是酶促转化生成甜菊糖苷d、m所需的反应,称为正常反应),还存在明显的副反应,即在c13-羟基方向或c19-羧基方向添加6号葡萄糖基(或4号葡萄糖基),二者与1号葡萄糖基形成β(1-6)糖苷键(或β(1-4)糖苷键)(本专利提及的副反应均指添加6号葡萄糖基(或4号葡萄糖基)的反应)(图1中b,c,d)。副反应将浪费酶催化资源和底物,形成明显的副产物,既影响了甜菊糖苷d和m的生成效率,还带来杂质,给获得高品质甜菊糖苷d、m纯品带来很大困难。
技术实现要素:
6.本发明所要解决的技术问题为:如何提高糖基转移酶osugt91c1在催化甜菊糖苷的c13-羟基或/和c19-羧基方向添加2号葡萄糖基的能力,另一方面,在提高催化能力的前提下消除副反应。
7.基于发明人独立发现的糖基转移酶osugt91c1三维空间结构,理性优化糖基转移酶osugt91c1原有氨基酸序列,发明人将第208位的苯丙氨酸改为甲硫氨酸(phe208met),得到新酶a。通过大肠杆菌蛋白表达系统,纯化制备新酶a,实施例验证新酶a可增强糖基转移酶osugt91c1在c13-羟基,特别是在c19-羧基位添加2号葡萄糖基的能力。
8.经前期研究发现,通过改变糖基转移酶osugt91c1第379位的苯丙氨酸为丙氨酸(phe379ala),可完全消除副反应。发明人综合新酶a(phe208met)催化能力得到增强的特点,组合phe208met和phe379ala两个位点的氨基酸改变,成功构建了一个既消除副反应又能增强目标催化能力(添加2号葡萄糖基)的新酶b(phe208met/phe379ala)。验证了新酶b(phe208met/phe379ala)既消除了添加6号葡萄糖基(形成β(1-6)糖苷键)的副反应,又对添加2号葡萄糖基的正常反应有约3-7倍的增强作用,可更高效和更加专一地参与甜菊糖苷的酶促转化。
9.进一步地研究发现,在上述突变的基础上,删除或改变糖基转移酶osugt91c1前端1-14个氨基酸(mdsgysssyaaaag)均不影响突变后的酶性质。说明前端的14个氨基酸具有冗余性,可以完全去除或改变。
10.本发明的技术方案为:糖基转移酶osugt91c1突变体,其氨基酸序列为以下a、b、c、d中的任一种:
11.a、seq id no.1所示;
12.b、seq id no.2所示;
13.c、seq id no.1所示氨基酸序列的基础上任意删除或改变第1-14位氨基酸;
14.d、seq id no.2所示氨基酸序列的基础上任意删除或改变第1-14位氨基酸。
15.seq id no.1所示氨基酸序列是在糖基转移酶osugt91c1氨基酸序列的基础上将第208位氨基酸由phe突变为met;seq id no.2所示氨基酸序列是在糖基转移酶osugt91c1氨基酸序列的基础上将第208位氨基酸由phe突变为met,且第379位氨基酸由phe突变为ala。
16.一种表达基因,编码上述所述的糖基转移酶osugt91c1突变体。
17.一种表达载体,含有上述所述表达基因。
18.携带上述所述表达载体的重组菌或细胞。
19.上述所述的糖基转移酶osugt91c1突变体在甜菊糖苷d或m及其他甜菊糖苷分子的酶促合成或转化过程的用途。
20.进一步地,所述糖基转移酶osugt91c1突变体在甜菊糖苷d或m及其他甜菊糖苷分子的酶促合成或转化过程是指在甜菊糖苷的c13-羟基或/和c19-羧基方向添加2号葡萄糖基,所述2号葡萄糖基是与1号葡萄糖基形成β(1-2)糖苷键的葡萄糖基,1号葡萄糖基特指直接与c13-羟基或c19-羧基形成β-糖苷键的葡萄糖基。
21.催化在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的底物不唯一,包括但不限于,rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a(rebaudioside a,cas 58543-16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应。
22.除了可在大肠杆菌中表达、纯化、制备天然状态osugt91c1或本发明的两个新酶,还可在其他生物系统、非生物系统、细胞、非细胞(cell-free)等其他表达体系中进行表达和制备;将这些消除了副反应的新酶,通过转基因或其他基因导入手段,引入生物体、非生物体、细胞、非细胞(cell-free)进行甜菊糖苷的酶促转化,以及在酶、酶固定化等体外体系,用于在甜菊糖苷添加2号葡萄糖基的酶促转化,包括制备但不限于甜菊糖苷d和m的酶促转化过程,达到消除副反应的目的。
23.本专利的副反应定义为,在甜菊醇或甜菊糖苷的c13-羟基,c19-羧基两个方向添加6号葡萄糖基(或4号葡萄糖基),与1号葡萄糖基形成β(1-6)糖苷键(或β(1-4)糖苷键)的副反应;本专利的正常反应的定义是在甜菊醇或甜菊糖苷的c13-羟基,c19-羧基两个方向添加2号葡萄糖基,与1号葡萄糖基形成β(1-2)糖苷键的反应。
24.利用了液相色谱-质谱联用验证了新酶a(phe208met)增强了催化添加2号葡萄糖基,特别是在c19-羧基位添加2号葡萄糖基的能力。新酶b(phe208met/phe379ala)在增强添加2号葡萄糖基能力的同时,无副反应的出现。通过液相色谱-质谱联用、荧光转化法等方法测定了两个新酶催化反应的米氏动力学参数,验证了新酶b(phe208met/phe379ala)既消除了添加6号葡萄糖基(形成β(1-6)糖苷键)的副反应,又对添加2号葡萄糖基的正常反应有约3-7倍的增强作用,可更高效和更加专一地参与甜菊糖苷的酶促转化。新酶b催化酶促转化,得到的添加了2号葡萄糖基的正常产物不含副产物,可用于合成甜菊糖苷d和m。
25.与现有技术相比,本发明具有以下有益效果:
26.本发明解决了水稻来源的糖基转移酶osugt91c1在甜菊糖苷转化中的酶学缺陷,同时显著提升了对目标催化反应的催化能力。新酶b(phe208met/phe379ala)可更为高效和专一地完成对甜菊糖苷添加2号葡萄糖基的正常酶促反应,用于转化获得高品质、高纯度的
健康甜味剂甜菊糖苷d和m。
附图说明
27.图1与本专利相关的部分甜菊糖苷结构,结构的简化表示及osugt91c1酶促转化生成甜菊糖苷d和m过程中正常反应和副反应的示意图
28.a部分甜菊糖苷的结构,包括甜菊醇、甜菊糖苷d和m的结构及结构的简化形式。甜菊醇用完整的化学结构表示,甜菊糖苷d和m用完整的化学结构和简化图两种方法表示,括号内为二者相比于蔗糖的甜度倍数。简化图中c13-羟基(用r1简化表示),c19-羧基(用r2简化表示)。葡萄糖基用六边形表示,每个葡萄糖的1-羟基用黑点标注,每个葡萄糖中间的数字代表是几号葡萄糖基,同时也表示该葡萄糖基与1号葡萄糖基形成的糖苷键的类型,如2代表β(1-2)糖苷键,3代表β(1-3)糖苷键,4代表β(1-4)糖苷键,6代表β(1-6)糖苷键。
29.b osugt91c1在甜菊糖苷底物c13-羟基(用r1简化表示)或(和)c19-羧基(用r2简化表示)两端催化添加2号葡萄糖基正常反应及添加6号葡萄糖基(或4号葡萄糖基)副反应的示意图。
30.c osugt91c1在甜菊糖苷底物c13-羟基(用r1简化表示)催化添加2号葡萄糖基正常反应及添加6号葡萄糖基(或4号葡萄糖基)副反应的示意图。
31.d osugt91c1在甜菊糖苷底物c19-羧基(用r2简化表示)催化添加2号葡萄糖基正常反应及添加6号葡萄糖基(或4号葡萄糖基)副反应的示意图。
32.图2新酶a(phe208met)、天然状态osugt91c1、新酶b(phe208met/phe379ala)sds-page分析
33.a新酶a(phe208met)的sds-page分析。m,marker;1,诱导后样品;2,菌体裂解液;3,菌体裂解液上清;4,菌体裂解液沉淀;5,ni柱流穿样品;6-12,ni柱洗脱蛋白样品。
34.b天然状态osugt91c1和新酶b(phe208met/phe379ala)的sds-page分析。m,marker;wt,天然状态osugt91c1;2,q柱流穿样品;3-7,q柱洗脱蛋白样品。两个图右侧箭头表示目的蛋白条带的位置。
35.图3天然状态osugt91c1和新酶a(phe208met)催化在底物rubu的c13-羟基(r1)和c19-羧基(r2)两端添加2号葡萄糖基,形成β(1-2)糖苷键正常酶促反应的对比
36.a,b天然状态osugt91c1、新酶a(phe208met)与rubu反应的液相分析。液相图从上至下的反应条件依次为:加0.25mg/ml酶反应0小时作为对照,0.05mg/ml酶反应2小时和18小时,0.25mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1在rubu的c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基的产物随时间和酶量的增加而增多,新酶a(phe208met)同样可在底物rubu的c13-羟基、c19-羧基两个方向添加2号葡萄糖基,而且可催化得到更多的在c19-羧基方向添加2号葡萄糖基的产物,具有增强的在c19-羧基方向的目标催化能力,最终得到在c13-羟基和c19-羧基两个方向都加有2号葡萄糖基的产物reb e也多于天然状态的osugt91c1;c,d天然状态osugt91c1和新酶a(phe208met)催化底物rubu的c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基,生成reb e的反应示意图,新酶a(phe208met)显示具有在c19-羧基方向增强的目标催化能力。
37.图4天然状态osugt91c1和新酶a(phe208met)催化在底物reb a的c19-羧基(r2)端添加2号葡萄糖基,形成β(1-2)糖苷键正常酶促反应的对比
38.a,b天然状态osugt91c1、新酶a(phe208met)与reb a反应的液相分析。液相图从上至下的反应条件依次为:加0.25mg/ml酶反应0小时作为对照,0.05mg/ml酶反应2小时和18小时,0.25mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1在reb a的c19-羧基(r2)端添加2号葡萄糖基,生成产物reb d随时间和酶量的增加而增多,而新酶a(phe208met)可明显率先将底物reb a完全消耗并生成目标产物reb d,具有增强的在c19-羧基方向的目标催化能力;c,d天然状态osugt91c1和新酶a(phe208met)催化底物reb a的c19-羧基(r2)端添加2号葡萄糖基,生成reb d的反应示意图,新酶a(phe208met)该反应得到增强。
39.图5天然状态osugt91c1和新酶b(phe208met/phe379ala)在底物stb的c13-羟基(r1)端添加6号(或4号)葡萄糖基副反应的情况对比
40.天然状态的osugt91c1(a)、新酶b(phe208met/phe379ala)(b)与底物stb反应的液相分析。液相图从上至下的反应条件依次为:加0.75mg/ml酶反应0小时作为对照,0.15mg/ml酶反应2小时和18小时,0.75mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1副反应产物随时间和酶量的增加而增多,而新酶b(phe208met/phe379ala)无副反应产物的生成。a液相图下方为osugt91c1催化stb反应生成副产物的示意图,b液相图下方为新酶b催化stb反应示意图,无副产物生成,无副反应的发生。
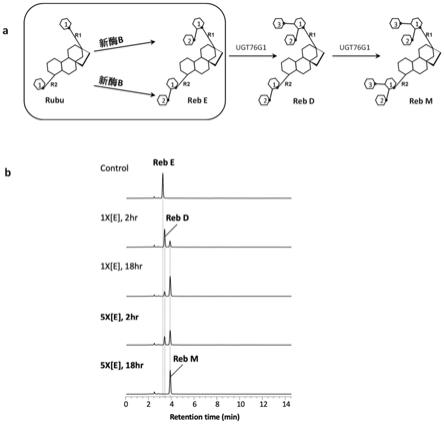
41.图6天然状态osugt91c1和新酶b(phe208met/phe379ala)催化在底物s13g的c13-羟基(r1)端添加2号葡萄糖基,形成β(1-2)糖苷键正常酶促反应,以及随后发生添加6号(或4号)葡萄糖基副反应的情况对比
42.天然状态的osugt91c1(a)、新酶b(phe208met/phe379ala)(b)与底物s13g反应的液相分析。液相图从上至下的反应条件依次为:加0.75mg/ml酶反应0小时作为对照,0.15mg/ml酶反应2小时和18小时,0.75mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1先催化在底物s13g的c13-羟基(r1)端添加2号葡萄糖基的正常酶促反应生成stb,然后在stb发生副反应,添加6号(或4号)葡萄糖基,生成副产物;与之相反,新酶b(phe208met/phe379ala)只催化在底物s13g的c13-羟基(r1)端添加2号葡萄糖基的正常反应生成stb,不催化副反应,无副产物生成;a液相图下方为天然状态osugt91c1催化s13g先催化正常反应生成stb,然后催化副反应产生副产物的示意图,b液相图下方为新酶b(phe208met/phe379ala)只催化s13g正常反应生成stb,不生成副产物的示意图。
43.图7天然状态osugt91c1和新酶b(phe208met/phe379ala)催化在底物rubu的c13-羟基(r1)端和c19-羧基(r2)端添加2号葡萄糖基,形成β(1-2)糖苷键正常酶促反应的对比
44.a,b天然状态osugt91c1、新酶b(phe208met/phe379ala)与rubu反应的液相分析。液相图从上至下的反应条件依次为:加0.25mg/ml酶反应0小时作为对照,0.05mg/ml酶反应2小时和18小时,0.25mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1在rubu的c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基的产物随时间和酶量的增加而增多,而新酶b(phe208met/phe379ala)可催化得到更多的在c19-羧基方向添加2号葡萄糖基的产物,具有增强的在c19-羧基方向的目标催化能力,而且最终得到在c13-羟基和c19-羧基两个方向都加有2号葡萄糖基的产物reb e也多于天然状态的osugt91c1;c,d天然状态osugt91c1和新酶b(phe208met/phe379ala)催化底物rubu的c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基,生成reb e的反应示意图,新酶b(phe208met/phe379ala)显示具
有更高的目标催化能力。
45.图8天然状态osugt91c1和新酶b(phe208met/phe379ala)催化在底物reb a的c19-羧基(r2)端添加2号葡萄糖基,形成β(1-2)糖苷键,生成reb d的正常酶促反应的对比
46.a,b天然状态osugt91c1、新酶b(phe208met/phe379ala)与reb a反应的液相分析。液相图从上至下的反应条件依次为:加0.25mg/ml酶反应0小时作为对照,0.05mg/ml酶反应2小时和18小时,0.25mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1在reb a的c19-羧基(r2)端添加2号葡萄糖基,生成产物reb d随时间和酶量的增加而增多,而新酶b(phe208met/phe379ala)可明显率先将底物反应完全,具有增强的在c19-羧基方向的目标催化能力;c,d天然状态osugt91c1和新酶b(phe208met/phe379ala)催化底物reb a的c19-羧基(r2)端添加2号葡萄糖基,生成reb d的反应示意图,其中新酶b(phe208met/phe379ala)的该反应得到增强。
47.图9利用新酶b(phe208met/phe379ala)催化获得的甜菊糖苷底物reb e(不产生副产物),通过糖基转移酶ugt76g1在c13-羟基或(和)c-19羧基方向继续添加3号葡萄糖基,获得甜菊糖苷d和m
48.a,以新酶b(phe208met/phe379ala)催化获得的甜菊糖苷底物reb e(不产生副产物)的基础上,再利用糖基转移酶ugt76g1转化形成reb d和reb m的反应示意图。方框显示两个新酶不发生副反应,只在c13-羟基或(和)c-19羧基方向正常添加2号葡萄糖基,形成甜菊糖苷e(reb e);然后糖基转移酶ugt76g1接力,继续催化在c13-羟基或(和)c-19羧基方向添加3号葡萄糖基,形成甜菊糖苷d和m(reb d和reb m)的过程。b,利用新酶b(phe208met/phe379ala)催化获得的甜菊糖苷底物reb e(不产生副产物),进一步利用糖基转移酶ugt76g1催化从reb e到reb d和reb m的转化过程。在液相图从上至下的反应条件依次为:加0.03mg/ml的糖基转移酶ugt76g1反应0小时作为对照,0.03mg/ml糖基转移酶ugt76g1反应2小时和18小时,0.15mg/ml糖基转移酶ugt76g1反应2小时和18小时。
49.图10新酶b(phe208met/phe379ala)去除第1-14位氨基酸后的截短体的sds-page分析;
50.新酶b(phe208met/phe379ala)和新酶b去除第1-14位氨基酸后的截短体的sds-page比较分析。m,marker;1,新酶b;2,新酶b的截短体从q柱流穿的样品;3-8,新酶b的截短体从q柱洗脱的样品
51.图11天然状态osugt91c1、新酶b去除第1-14位氨基酸后的截短体催化在底物rubu的c13-羟基(r1)和(或)c19-羧基(r2)端分别添加2号葡萄糖基,形成β(1-2)糖苷键正常酶促反应的对比;
52.天然状态的osugt91c1(a)、新酶b去除第1-14位氨基酸后的截短体(b)与rubu反应的液相分析比较。液相图从上至下的反应条件依次为:加0.25mg/ml酶反应0小时作为对照,0.05mg/ml酶反应2小时和18小时,0.25mg/ml酶反应2小时和18小时。显示天然状态的osugt91c1在rubu的c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基的产物随时间和酶量的增加而增多,而新酶b截短体具有同样的酶促催化反应,且同新酶b一样,具有比天然状态osugt91c1更好的催化能力。c,天然状态osugt91c1和新酶b截短体可进行同样催化在底物rubu的c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基的正常目标反应,生成reb e的反应示意图,进一步验证第1-14位氨基酸的冗余性,可以完全去除或改变。
具体实施方式
53.下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为从商业渠道购买得到的。
54.一、天然状态osugt91c1蛋白表达载体的构建
55.(1)获取osugt91c1的氨基酸序列(ncbi reference sequence:xp_015629141.1),根据蛋白表达系统的要求对osugt91c1编码密码子进行优化和合成。发明人根据大肠杆菌的表达体系优化后的编码密码子如下。由于密码子的兼并性,密码子优化的选择可有多种。
56.为了防止通过替换osugt91c1的非重要氨基酸序列,达到规避本专利权利要求的目的,本专利认为只要翻译后的蛋白氨基酸序列和osugt91c1的氨基酸序列相同度在50%及以上,则认为属于osugt91c1。
57.不管是在osugt91c1的氨基酸序列还是空间结构上,通过改变同208和379位氨基酸等效的位点或其他位点(由于氨基酸冗余性的原因,通过去除第1-14位的一个或任意个氨基酸,导致第208和379位氨基酸不一定一直是第208和379位的编号),达到提升osugt91c1添加2号葡萄糖基的正常反应或(和)消除osugt91c1副反应的目的,都属于本专利的权利要求。
58.osugt91c1的氨基酸序列(加粗的氨基酸是本专利进行改动的氨基酸位点,划线部分是人为添加的氨基酸序列,其中le是为了方便连接进入大肠杆菌表达载体pet21b,hhhhhh(6个组氨酸标签)是为了方便后期的纯化):
[0059][0060]
osugt91c1密码子优化后的核苷酸序列(划线序列为上述人为添加氨基酸序列的编码区):
[0061]
atggatagcggttatagtagcagttatgccgcagccgccggcatgcatgttgtgatttgcccgtggctggcctttggtcatctgctgccgtgcttagacctggcccagcgtctggccagccgtggtcaccgtgttagctttgtgagcaccccgcgtaatatcagccgtctgccgccggttcgtccggcattagccccgctggtggcatttgtggccttaccgctgccgcgtgttgagggtctgcctgatggcgccgaaagtaccaacgacgtgccgcatgaccgcccggatatggtggagctgcatcgtcgcgcctttgatggtctggcagccccgtttagcgagtttctgggcacagcctgcgccgattgggtgatcgttgacgtgtttcatcactgggcagccgcagccgccctggaacataaagttccgtgcgcaatgatgctgctgggtagcgcccacatgattgccagcattgccgatcgtcgcctggaacgcgcagagaccgaaagcccggcagcagcaggtcaaggtcgtcctgccgcagccccgacctttgaagtggcccgcatgaaactgatccgtaccaaaggtagtagcggcatgagcctggccgaacgctttagcctgaccctgagccgcagtagcctggtggttggtcgcagttgtgtggaattcgagccggaaacagtgccgctgctgagcaccctgcgcggcaaaccgatcacctttctgggcctgatgccgccgtt
acatgaaggccgtcgtgaagatggtgaagatgccacagtgcgttggctggatgcacagccggccaaaagcgttgtgtacgttgccctgggtagcgaagttcctctgggtgtggaaaaggtgcacgaactggcactgggtctggaactggccggtacccgcttcctgtgggccttacgtaaacctaccggtgttagcgatgccgatctgctgccggcaggttttgaggaacgtacccgtggtcgcggtgttgtggcaacacgctgggttccgcagatgagcattctggcccatgccgccgtgggtgcctttctgacccattgtggctggaatagcaccatcgaaggcctgatgttcggccatcctctgatcatgctgcctatcttcggtgatcagggtccgaacgcacgcctgattgaagcaaagaatgccggtctgcaggtggcacgtaacgatggcgacggtagcttcgatcgtgaaggcgttgccgccgcaattcgcgccgttgcagttgaagaagagagcagcaaggtgttccaggccaaagccaaaaaactgcaggagatcgtggccgatatggcatgccatgagcgctacatcgatggcttcatccagcagctgcgcagctataaagatctcgagcaccaccaccaccaccac
[0062]
(2)对合成得到的osugt91c1编码区用表1所列的引物进行扩增,扩增片段的5’和3’端将分别带上nde i和xho i的限制酶位点。
[0063]
表1 pet21b-osugt91c1的引物序列
[0064][0065]
(3)用nde i和xho i对pet21b表达载体和步骤2的osugt91c1编码区扩增片段进行双酶切,将osugt91c1编码区用t4连接酶连接进pet21b载体的nde i和xho i酶切位点之间,组成在大肠杆菌可表达糖基转移酶osugt91c1的表达载体pet21b-osugt91c1。
[0066]
(4)将连接产物全部转入100μle.coli dh5α感受态细胞中,挑取平板上的阳性单克隆菌落接种于10ml的lb培养基中,200rpm 37℃培养12-16小时后提取质粒,测序验证表达质粒的正确性,完成天然状态osugt91c1糖基转移酶表达载体的构建。
[0067]
二、为了增强添加2号葡萄糖基的正常反应,构建新酶a(phe208met)的表达载体;为了既提升酶促添加2号葡萄糖基的目标催化活性,又消除副反应,构建新酶b(phe208met/phe379ala)的表达载体
[0068]
1、新酶a(phe208met)表达载体的构建
[0069]
(1)以天然状态pet21b-osugt91c1为模板,设计新酶a(phe208met)和新酶b(phe208met/phe379ala)所使用的定点突变引物如表2。
[0070]
表2突变体引物序列如下:(下划线部分表示突变位点)
[0071][0072]
(2)用ddh2o溶解表2的引物,稀释引物浓度为10μm。用phe208met的一对突变引物以pet21b-osugt91c1为模板进行pcr扩增,体系同为:dntp 4μl,5
×
ps buffer 10μl,上下游引物各2μl,模板1μl(约10ng),pcr扩增酶primer star 0.5μl,剩余用ddh2o补齐至50μl。混匀后用pcr扩增,扩增程序:98℃预变性2min,98℃变性30s,69℃退火30s,72℃延伸8min,
扩增20个循环,72℃再延伸10min,最后4℃保存。
[0073]
(3)从上述扩增产物取出10μl进行琼脂糖胶验证pcr的扩增效果,剩余的40μl体系里各加1μldpni酶,37℃孵育1-2小时后取10μl分别转入100μle.coli dh5α感受态细胞中,冰浴30min,42℃热激2min,再冰浴3min后加入新鲜lb 300μl,200rpm 37℃摇床孵育1小时后,分别取150μl均匀涂布于amp抗性的固体lb平板上,37℃静置培养过夜。
[0074]
(4)挑取两个平板上的单克隆菌落接种于10ml的lb培养基中,200rpm 37℃培养12-16小时后提取质粒,dna测序验证突变位点phe208met的正确性,编码的氨基酸序列如seq id no.1所示。
[0075]
2、新酶b(phe208met/phe379ala)表达载体的构建
[0076]
(1)获得上述2.2.2.1第4步经过测序验证的新酶a(phe208met)的表达载体为模板,进行新酶b(phe208met/phe379ala)表达载体的构建;
[0077]
(2)用ddh2o溶解表2的引物,稀释引物浓度为10μm。用phe379ala的一对突变引物以新酶a(phe208met)的表达载体为模板进行pcr扩增,体系同上:dntp 4μl,5
×
ps buffer 10μl,上下游引物各2μl,模板1μl(约10ng),pcr扩增酶primer star 0.5μl,剩余用ddh2o补齐至50μl。混匀后用pcr扩增,扩增程序:98℃预变性2min,98℃变性30s,68℃退火30s,72℃延伸8min,扩增20个循环,72℃再延伸10min,最后4℃保存。
[0078]
(3)从上述扩增产物取出10μl进行琼脂糖胶验证pcr的扩增效果,剩余的40μl体系里各加1μldpni酶,37℃孵育1-2小时后取10μl分别转入100μle.coli dh5α感受态细胞中,冰浴30min,42℃热激2min,再冰浴3min后加入新鲜lb 300μl,200rpm 37℃摇床孵育1小时后,分别取150μl均匀涂布于amp抗性的固体lb平板上,37℃静置培养过夜。
[0079]
(4)挑取两个平板上的单克隆菌落接种于10ml的lb培养基中,200rpm 37℃培养12-16小时后提取质粒,dna测序验证两个突变位点phe208met和phe379ala的正确性,编码的氨基酸序列如seq id no.2所示。
[0080]
三、天然状态osugt91c1和新酶a,新酶b的诱导表达和纯化
[0081]
(1)将上述三种酶对应的表达质粒转化至e.coli bl21(de3)表达菌株,第二天挑取单克隆接种于10ml新鲜的lb培养基中,200rpm 37℃培养过夜后加入终浓度为8%的甘油保种,该菌种可在-80℃长期保存。
[0082]
(2)挑取上一步保存的e.coli bl21(de3)甘油菌接种于含amp 50μg/ml的100ml新鲜lb培养基中,200rpm 37℃过夜培养。第二天以1%的接种比例接种于含amp 50μg/ml的1l新鲜lb培养基,180rpm 37℃培养至od600=1.0后,菌液降温至16℃,添加终浓度为0.5mm的iptg,160rpm 16℃
–
20℃诱导表达18小时。
[0083]
(3)完成诱导表达后,4000rpm离心15min弃上清,收集菌体。菌体用重悬缓冲液(20mm tris-hcl buffer ph 7.8,0.5m nacl,30mm imidazole)重悬后,用高压破碎仪在1000bar压力下反复破碎3次。13500rpm 4℃离心60min,离心后的上清上样到nta-ni柱,用上述重悬缓冲液洗去非特异性结合的杂蛋白,带组氨酸标签的目的蛋白可被洗脱缓冲液(20mm tris-hcl buffer ph 7.8,0.5m nacl,250mm imidazole)洗脱下来。洗脱下来的蛋白进一步置换到20mm hepes buffer ph 7.2,50mm nacl里,经液氮速冻后,可长期保存于-80℃。上述步骤可纯化得到天然状态的osugt91c1、新酶a、新酶b,并通过sds-page检测纯度(图2)。
16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应。
[0092]
2、酶促产物分析,表明新酶b(phe208met/phe379ala)消除了添加6号葡萄糖基(或4号葡萄糖基)的副反应,但不影响在c13-羟基和c19-羧基方向添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的催化能力
[0093]
(1)以osugt91c1的底物stb(steviolbioside,cas 41093-60-1)为实施例,天然状态的osugt91c1可分别在底物stb上催化添加6号葡萄糖基(或4号葡萄糖基)的副反应(图5中a),天然状态的osugt91c1副反应产物随时间和酶量的增加而增多,而新酶b(phe208met/phe379ala)无副反应产物的生成,已经消除添加6号葡萄糖基(或4号葡萄糖基)的副反应(图5中b)。
[0094]
副反应的实施例如下:在20℃-40℃,200μl反应体系包括:1mm udp-glucose,20mm tris-hcl缓冲液ph 7.2,浓度分别为0.15mg/ml(1
×
)和0.75mg/ml(5
×
)的酶样品(天然状态或者新酶b(phe208met/phe379ala)),0.3mm stb。通过加入酶样品启动反应,分别于0,2,18小时取样60μl,与等体积的正丁醇混合涡旋振荡,终止反应并萃取对应的酶促反应产物,室温17000rpm离心10min,室温静置1min,取上层正丁醇的萃取相50μl真空干燥后,用等体积的25%乙腈重悬后,利用hplc检测酶促产物。本实施例所列的反应条件不唯一,只要能使osugt91c1发生酶促反应,都可得到同样的结果。检测手段也不唯一,只要能区分每个酶促产物,可得到相同的检测结果。
[0095]
天然状态的osugt91c1催化副反应的底物不唯一,包括但不限于对以下甜菊糖苷底物发生副反应,stb(steviolbioside,cas 41093-60-1),reb e(rebaudioside e,cas 63279-14-1)等,只要在c13-羟基或c-19羧基方向存在1号和2号葡萄糖基,但不存在3号葡萄糖基(3号葡萄糖基指与1号葡萄糖基形成β(1-3)糖苷键的葡萄糖基),就可发生在对应的c13-羟基或c-19羧基方向添加6号葡萄糖基(或4号葡萄糖基)的副反应。
[0096]
(2)以osugt91c1的底物s13g(steviol-13-o-monoglucoside,cas 60129-60-4)为实施例,天然状态的osugt91c1先催化在底物s13g的c13-羟基(r1)端添加2号葡萄糖基的正常酶促反应生成stb,然后对stb发生副反应,添加6号(或4号)葡萄糖基,生成副产物(图6中a);与之相反,新酶b(phe208met/phe379ala)只催化在底物s13g的c13-羟基(r1)端添加2号葡萄糖基的正常反应生成stb,不催化副反应,无副产物生成(图6中b)。
[0097]
实施例的反应条件如下:在20℃-40℃,200μl反应体系包括:1mm udp-glucose,20mm tris-hcl缓冲液ph 7.2,浓度分别为0.15mg/ml(1
×
)和0.75mg/ml(5
×
)的酶样品(天然状态或者新酶b(phe208met/phe379ala)),0.3mm底物s13g。通过加入酶样品启动反应,分别于0,2,18小时取样60μl,与等体积的正丁醇混合涡旋振荡,终止反应并萃取对应的酶促反应产物,室温17000rpm离心10min,室温静置1min,取上层正丁醇的萃取相50μl真空干燥后,用等体积的25%乙腈重悬后,利用hplc检测酶促产物。本实施例所列的反应条件不唯一,只要能使osugt91c1发生酶促反应,都可得到同样的结果。检测手段也不唯一,只要能区分每个酶促产物,可得到相同的检测结果。
[0098]
天然状态的osugt91c1和新酶b(phe208met/phe379ala)催化在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的底物不唯一,包括但不限于,rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a
(rebaudioside a,cas 58543-16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应;天然状态的osugt91c1催化副反应的底物不唯一,包括但不限于对以下甜菊糖苷底物发生副反应,stb(steviolbioside,cas 41093-60-1),reb e(rebaudioside e,cas 63279-14-1)等,只要在c13-羟基或c-19羧基方向存在1号和2号葡萄糖基,但不存在3号葡萄糖基(3号葡萄糖基指与1号葡萄糖基形成β(1-3)糖苷键的葡萄糖基),就可发生在对应的c13-羟基或c-19羧基方向添加6号葡萄糖基(或4号葡萄糖基)的副反应。
[0099]
3、酶促产物分析,表明新酶b(phe208met/phe379ala)提升了在c13-羟基,特别是c19-羧基方向添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的催化能力
[0100]
(1)以osugt91c1的底物rubu(rubusoside,cas 64849-39-4)为实施例,天然状态的osugt91c1可在底物rubu的c13-羟基、c19-羧基两个方向添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)(正常反应)(图7中a),而新酶b(phe208met/phe379ala)同样可在底物rubu的c13-羟基、c19-羧基两个方向添加2号葡萄糖基,而且可催化得到更多的在c19-羧基方向添加2号葡萄糖基的产物,具有增强的在c19-羧基方向的目标催化能力,最终得到在c13-羟基和c19-羧基两个方向都加有2号葡萄糖基的产物reb e(rebaudioside e,cas 63279-14-1)也明显多于天然状态的osugt91c1(图7中b)。
[0101]
实施例的反应体系如下:在20℃-40℃,200μl反应体系包括:1mm udp-glucose,20mm tris-hcl缓冲液ph 7.2,浓度分别为0.05mg/ml(1
×
)和0.25mg/ml(5
×
)的酶样品(天然状态或者新酶b(phe208met/phe379ala)),0.3mm底物rubu。通过加入酶样品启动反应,分别于0,2,18小时取样60μl,与等体积的正丁醇混合涡旋振荡,终止反应并萃取对应的酶促反应产物,室温17000rpm离心10min,室温静置1min,取上层正丁醇的萃取相50μl真空干燥后,用等体积的25%乙腈重悬后,利用hplc检测酶促产物。本实施例所列的反应条件不唯一,只要能使osugt91c1发生酶促反应,都可得到同样的结果。检测手段也不唯一,只要能区分每个酶促产物,可得到相同的检测结果。
[0102]
天然状态的osugt91c1和新酶b(phe208met/phe379ala)催化在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的底物不唯一,包括但不限于,rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a(rebaudioside a,cas 58543-16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应。
[0103]
(2)以osugt91c1的底物reb a(rebaudioside a,cas 58543-16-1)为实施例,天然状态的osugt91c1可在底物reb a的c19-羧基方向添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)(正常反应)(图8中a),而新酶b(phe208met/phe379ala)可明显率先将底物reb a完全消耗并生成目标产物reb d(rebaudioside d,cas 63279-13-0),具有增强的在c19-羧基方向的目标催化能力,提高在c19-羧基方向添加2号葡萄糖基形成对应产物的能力(图8中b)。
[0104]
实施例的反应体系如下:在20℃-40℃,200μl反应体系包括:1mm udp-glucose,20mm tris-hcl缓冲液ph 7.2,浓度分别为0.05mg/ml(1
×
)和0.25mg/ml(5
×
)的酶样品(天然状态或者新酶b(phe208met/phe379ala)),0.3mm底物reb a。通过加入酶样品启动反应,
分别于0,2,18小时取样60μl,与等体积的正丁醇混合涡旋振荡,终止反应并萃取对应的酶促反应产物,室温17000rpm离心10min,室温静置1min,取上层正丁醇的萃取相50μl真空干燥后,用等体积的25%乙腈重悬后,利用hplc检测酶促产物。本实施例所列的反应条件不唯一,只要能使osugt91c1发生酶促反应,都可得到同样的结果。检测手段也不唯一,只要能区分每个酶促产物,可得到相同的检测结果。
[0105]
天然状态的osugt91c1和新酶b(phe208met/phe379ala)催化在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的底物不唯一,包括但不限于,rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a(rebaudioside a,cas 58543-16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应。
[0106]
4、通过荧光转化的方法,测定新酶a(phe208met)和新酶b(phe208met/phe379ala)催化在甜菊糖苷底物添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)正常反应的速度。
[0107]
新酶a(phe208met)增强了催化正常所需的在甜菊糖苷底物添加第2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的反应速度,是天然状态osugt91c1活性的2倍以上;新酶b(phe208met/phe379ala)消除添加6号(或4号)葡萄糖基副反应的同时,增强了催化正常所需的在甜菊糖苷底物添加第2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的反应速度,是天然状态osugt91c1活性的3-7倍(表3)。
[0108]
表3天然状态osugt91c1、新酶a(phe208met)、新酶b(phe208met/phe379ala)催化在底物c13-羟基(r1)和c19-羧基(r2)端分别添加2号葡萄糖基(正常所需的催化反应)的动力学参数
[0109][0110]
(1)该测定直接利用商品化的糖基转移酶活性试剂盒完成,实施例使用promega公司的udp-glo
tm glycosyltransferase assay试剂盒,其他可用于糖基转移酶反应速度检测方法可得到同样结果。
[0111]
promega公司的udp-glo
tm glycosyltransferase assay试剂盒用于检测以udp-glucose为糖基供体的葡萄糖基转移酶反应速度,osugt91c1及本发明涉及的两个突变新酶均适用。
[0112]
在葡萄糖基转移酶的作用下,糖基供体udp-glucose将葡萄糖转移给底物后生成udp。该检测方法将生成udp等物质的量(1:1)转化为atp,atp可使荧光素酶发出定量的荧光,因此通过测定荧光量可检测酶促糖基转移反应生成udp的量。当生成udp在0-25μm浓度
范围内,产生的荧光强度和udp的摩尔浓度成线性关系。根据udp浓度和荧光强度对应关系的标准曲线,计算udp的生成量,可转化为糖基转移酶的催化反应速度。
[0113]
(2)配制udp不同浓度的一系列溶液,根据上述转化的反应,测定对应的荧光强度。天然状态osugt91c1及本发明涉及的两个新酶,以不同底物检测各种催化反应的速度及酶动力学常数等信息,用于评价本发明的新酶a(phe208met)和新酶b(phe208met/phe379ala)在促进添加2号葡萄糖基正常反应上的效果。利用rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a(rebaudioside a,cas 58543-16-1)为底物分别检测新酶a(phe208met)和新酶b(phe208met/phe379ala)在c13-羟基,c19-羧基添加2号葡萄糖基的催化能力,并和天然状态osugt91c1的催化能力进行比较。天然状态的osugt91c1及新酶a(phe208met)、新酶b(phe208met/phe379ala)催化在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的底物不唯一,包括但不限于,rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a(rebaudioside a,cas 58543-16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应。
[0114]
(3)天然状态osugt91c1与新酶a(phe208met)和新酶b(phe208met/phe379ala)针对不同底物的酶动力学常数表3所示,新酶a(phe208met)增强了催化正常所需的在甜菊糖苷底物添加第2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的反应速度,是天然状态osugt91c1活性的2倍以上;新酶b(phe208met/phe379ala)消除添加6号(或4号)葡萄糖基副反应的同时,增强了催化正常所需的在甜菊糖苷底物添加第2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)的反应速度,是天然状态osugt91c1活性的3-7倍(表3)。
[0115]
五、新酶b(phe208met/phe379ala)既无副反应的发生又提升了目标催化能力,可催化完成在甜菊糖苷底物c13-羟基或(和)c-19羧基方向添加2号葡萄糖基的正常反应,生成包括reb e(rebaudioside e,cas 63279-14-1)的一系列甜菊糖苷产物。在此条件下,利用糖基转移酶ugt76g1在c13-羟基或(和)c-19羧基方向继续添加3号葡萄糖基,则可获得甜菊糖苷d和m(rebaudioside d,cas 63279-13-0和rebaudioside m,cas 1220616-44-3)(图9)。
[0116]
六、新酶a(phe208met)、新酶b(phe208met/phe379ala)的氨基酸序列具有冗余性,可以去除或改变第1-14位的任一氨基酸。
[0117]
以新酶b(phe208met/phe379ala)为实施例,对新酶b去除第1-14位的全部氨基酸(mdsgysssyaaaag)得到新酶b截短体,该截短体仍能正常表达、纯化,并显示出在甜菊糖苷底物c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的活性。
[0118]
1、为了验证第1-14位氨基酸(mdsgysssyaaaag)的冗余性,在新酶b(phe208met/phe379ala)的表达载体上构建去除第1-14位的全部氨基酸(mdsgysssyaaaag)(新酶b截短体)的表达载体
[0119]
(1)以新酶b(phe208met/phe379ala)的表达质粒为模板,设计去除第1-14位的全部氨基酸的截短体的引物如表4。
[0120]
表4突变体引物序列如下:
[0121][0122]
(2)用ddh2o溶解表4的引物,稀释引物浓度为10μm。用去除1-14-f和去除1-14-r的一对引物分别以新酶b(phe208met/phe379ala)的表达质粒为模板进行pcr扩增,体系同为:dntp 4μl,5
×
ps buffer 10μl,上下游引物各2μl,模板1μl(约10ng),pcr扩增酶primer star 0.5μl,剩余用ddh2o补齐至50μl。混匀后用pcr扩增,扩增程序:98℃预变性2min,98℃变性30s,69℃退火30s,72℃延伸8min,扩增20个循环,72℃再延伸10min,最后4℃保存。
[0123]
(3)从上述扩增产物各取出10μl进行琼脂糖胶验证pcr的扩增效果,剩余的40μl体系里各加1μl dpni酶,37℃孵育1-2小时后取10μl分别转入100μl e.coli dh5α感受态细胞中,冰浴30min,42℃热激2min,再冰浴3min后加入新鲜lb 300μl,200rpm37℃摇床孵育1小时后,分别取150μl均匀涂布于amp抗性的固体lb平板上,37℃静置培养过夜。
[0124]
(4)挑取两个平板上的单克隆菌落接种于10ml的lb培养基中,200rpm 37℃培养12-16小时后提取质粒,dna测序验证新酶b截短体的表达质粒,成功去除了第1-14位的全部氨基酸,除了第1-14位氨基酸被去除,其余位点氨基酸序列同seq id no.2维持一致。
[0125]
2、新酶b(phe208met/phe379ala)去除第1-14位的全部氨基酸后截短体的诱导表达和纯化
[0126]
(1)将对应的表达质粒转化至e.coli bl21(de3)表达菌株,第二天挑取单克隆接种于10ml新鲜的lb培养基中,200rpm 37℃培养过夜后加入终浓度为8%的甘油保种,该菌种可在-80℃长期保存。
[0127]
(2)挑取上一步保存的e.coli bl21(de3)甘油菌接种于含amp 50μg/ml的100ml新鲜lb培养基中,200rpm 37℃过夜培养。第二天以1%的接种比例接种于含amp 50μg/ml的1l新鲜lb培养基,180rpm 37℃培养至od600=1.0后,菌液降温至16℃,添加终浓度为0.5mm的iptg,160rpm 16℃
–
20℃诱导表达18小时。
[0128]
(3)完成诱导表达后,4000rpm离心15min弃上清,收集菌体。菌体用重悬缓冲液(20mm tris-hcl buffer ph 7.8,0.5m nacl,30mm imidazole)重悬后,用高压破碎仪在1000bar压力下反复破碎3次。13500rpm 4℃离心60min,离心后的上清上样到nta-ni柱,用上述重悬缓冲液洗去非特异性结合的杂蛋白,带组氨酸标签的目的蛋白可被洗脱缓冲液(20mm tris-hcl buffer ph 7.8,0.5m nacl,250mm imidazole)洗脱下来。洗脱下来的蛋白进一步置换到20mm hepes buffer ph 7.2,50mm nacl里,经液氮速冻后,可长期保存于-80℃。上述步骤可纯化得到新酶b的截短体,并通过sds-page检测纯度(图10)。由图10可见,当新酶b去除第1-14位氨基酸的截短体可以进行正常的表达和纯化。由于新酶a和新酶b具有同样的第1-14位氨基酸,对新酶b完全去除第1-14位氨基酸的实施例,显示第1-14位氨基酸具有冗余性。同理,新酶a的第1-14位氨基酸也具有冗余性。
[0129]
3、新酶b的截短体不影响在甜菊糖苷底物的c13-羟基和c-19羧基两个方向添加第2号葡萄糖基的正常反应,其活性和新酶b一样,比天然转态糖基转移酶osugt91c1的催化活性更高,进一步显示第1-14位氨基酸在新酶a和新酶b中的冗余性。
[0130]
(1)以甜菊糖苷底物rubu(rubusoside,cas 64849-39-4)为实施例,天然状态的
osugt91c1可在底物rubu的c13-羟基、c19-羧基两个方向添加2号葡萄糖基(与1号葡萄糖基形成β(1-2)糖苷键)(正常反应)(图11中a),新酶b的截短体同样可在底物rubu的c13-羟基、c19-羧基两个方向添加2号葡萄糖基(图11中b)。
[0131]
(2)实施例的反应体系如下:在20℃-40℃,200μl反应体系包括:1mm udp-glucose,20mm tris-hcl缓冲液ph 7.2,浓度分别为0.05mg/ml(1
×
)和0.25mg/ml(5
×
)的酶样品(天然状态或者新酶b截短体),0.3mm底物rubu。通过加入酶样品启动反应,分别于0,2,18小时取样60μl,与等体积的正丁醇混合涡旋振荡,终止反应并萃取对应的酶促反应产物,室温17000rpm离心10min,室温静置1min,取上层正丁醇的萃取相50μl真空干燥后,用等体积的25%乙腈重悬后,利用hplc检测酶促产物。本实施例所列的反应条件不唯一,只要能使osugt91c1发生酶促反应,都可得到同样的结果。检测手段也不唯一,只要能区分每个酶促产物,可得到相同的检测结果。
[0132]
(3)天然状态的osugt91c1和新酶b截短体催化在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应的底物不唯一,包括但不限于,rubu(rubusoside,cas 64849-39-4)、s13g(steviol-13-o-monoglucoside,cas 60129-60-4)及reb a(rebaudioside a,cas 58543-16-1)等甜菊糖苷底物,只要在c13-羟基或c-19羧基方向存在1号葡萄糖基,但不存在3号葡萄糖基,就可发生在c13-羟基或c-19羧基方向添加2号葡萄糖基的正常反应。