基于游离dna的基因组癌变信息检测系统和检测方法
技术领域
1.本发明涉及基因组癌变信息检测领域,尤其涉及一种基于游离dna的基因组癌变信息检测系统和检测方法。
背景技术:
2.癌症的早筛、早诊可以为及时治疗提供可能,从而降低癌症的死亡率。传统的肿瘤诊断技术主要为影像学检查,例如胃镜、结肠镜检查,作为侵入性检测手段可能会对患者造成创伤,且检测灵敏度受限于肿瘤发展阶段,只能发现直径1 cm以上的肿瘤病灶,发现时基本到了中晚期。病理学组织活检是癌症诊断的金标准,但检取样困难,且由于肿瘤的异质性往往难以做到取样完全,不利于诊断分型,又容易导致并发症。液体活检技术,特别是基于检测血浆中的游离dna(cell-free dna,cfdna)中肿瘤来源的游离肿瘤dna(circulating tumor dna, ctdna)的生物标志物信号的检测技术,近年来作为一种非侵入性肿瘤检测手段被广泛应用于肿瘤诊断、病情追踪、复发监测等。相比较于传统影像学方法,液体活检技术对于早期肿瘤有更高的检测灵敏度,且可以实现对多癌种的同时检测,具有作为一种针对普通人群的常规癌症筛查手段的潜力。
3.ctdna来源于坏死的、凋亡的、循环中的肿瘤细胞以及肿瘤细胞分泌的外排体,携带着肿瘤细胞的遗传和表观遗传特征。dna甲基化是真核细胞中的重要表观修饰方式,即在dna甲基化转移酶(dna methyltransferases, dnmts)的作用下使cpg岛的胞嘧啶(cytosine)转变为5
’‑
甲基胞嘧啶(5-mc)。dna甲基化状态的改变是肿瘤发生、发展过程中的标志性事件之一,在肿瘤早期便在基因组广泛发生。人类基因启动子区的cpg岛在癌症中常发生高甲基化现象,可能会沉默某些抑癌基因的表达;同时癌症基因组常呈现大范围的去甲基化状态,可能会导致重复序列区域的激活或者染色体重排。
4.通过检测血浆cfdna甲基化状态的改变可以灵敏的检测微弱的ctdna信号。人类基因组大于3g,出于测序成本的考虑,目标区域捕获测序是目前最常用的甲基化检测手段,但是其性能受限于对癌种特异性目标区域的筛选,需要前期对癌症和匹配的癌旁组织进行高深度全基因组甲基化测序分析来选择差异甲基化位点。因而,该技术路径的一大瓶颈为各癌种高质量组织样本的获得,且差异甲基化位点的筛选和验证过程较为繁琐。
5.除了甲基化状态的改变,癌症病人的cfdna的片段化特征,包括全基因组各区域不同长度的片段的比例、片段末端序列等,也呈现出与健康人的差异,近年来作为另一种灵敏的ctdna的表观遗传生物标志物被广泛开发用于多个癌种的检测(“片段组学”)。此外,拷贝数变异(copy number variation,cnv)是各种癌症中常见的遗传特征改变,也被广泛应用于对ctdna信号的检测。
6.传统的甲基化测序技术利用重亚硫酸盐将非甲基化的胞嘧啶(c)脱氨转变成尿嘧啶(u),该反应的高温和高ph环境会引起dna分子的严重降解,从而丢失原始的dna片段特征。
技术实现要素:
7.仍然需要开发针对基于游离dna构建的单个测序文库能够同时分析包括甲基化、片段化特征、拷贝数变异等特征,能够更准确、更灵敏、更廉价、更简便地检测基因组癌变信息的系统和方法,同时用于多种癌症的早期、灵敏、准确筛查。
8.本发明是基于发明人的下列发现而完成的:发明人首次发现,通过对血浆cfdna(cell-free dna)进行酶法处理,使其中的5-甲基胞嘧啶(5-mc)转化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),非甲基化胞嘧啶(c)转化为尿嘧啶(u),可获得测序文库,同时用于全基因组的甲基化、片段化(例如从片段长度系数分析和末端基序(motif)分析两个维度)、染色体不稳定性分析(拷贝数变异),同时对多种癌症进行早期、灵敏、准确的筛查。
9.本发明提供了一种低成本的能对血浆cfdna同时进行全基因组甲基化、片段化以及拷贝数变异分析的文库构建方法及分析模型进行癌症的液体活检筛查,该方法适用于低起始量cfdna,无需进行目标区域捕获从而简化技术流程。进一步地,本发明可以可选地通过对上述各维度癌症特征的整合分析进一步提高癌症筛查的检测灵敏度和准确性。
10.一方面,本文提供了一种基于游离dna(cell free dna,cfdna的基因组癌变信息检测系统,包括:文库构建装置,通过利用酶使待测样品中游离dna(例如血浆中的游离dna)中的5-甲基胞嘧啶(5-mc)转化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),非甲基化胞嘧啶(c)转化为尿嘧啶(u),用于构建文库;测序装置,用于对所构建的文库进行测序;信息分析装置,其包括以下一个或多个模块:甲基化分析模块,用于分析游离dna的甲基化信息,片段长度系数分析模块,用于分析游离dna的片段化信息,末端基序分析模块,用于分析游离dna的片段化信息,染色体不稳定性分析模块,用于分析染色体的拷贝数变异信息。
11.在一些实施方案中,所述信息分析装置还包括整合分类模块,用于将所述甲基化分析模块、片段长度系数分析模块、末端基序分析模块和/或染色体不稳定性分析模块所获得的信息进行整合。
12.在一些实施方案中,所述甲基化分析模块是md-knn分析模块,通过非重叠滑窗方法将人参考基因组化分为区间(即bin,例如1mb大小),计算每个区间的所有cpg位点中甲基化位点的比例,即甲基化密度md(methylation density)值,通过knn(k
‑ꢀ
nearest neighbor,k临近法)模型计算癌变可能性的预测值k。
13.在一些具体实施方案中,所述片段长度系数分析模块是fsi-svm分析模块,通过非重叠滑窗方法将人参考基因组化分为区间(例如5mb大小),计算每个区间的短片段(例如101-167bp)和长片段(例如170-250bp)数目的比例,得到每个样本的片段长度系数fsi(fragment size index)值,通过svm(support vector machine,支持向量机)模型计算癌变可能性的预测值f。
14.在一些实施方案中,所述末端基序分析模块是motif-svm分析模块,计算样本的片段的5’末端4-mer基序序列的占比,通过svm模型计算癌变可能性的预测值s。
15.在一些实施方案中,所述染色体不稳定性分析模块是cin-pascore分析模块,计算
样本的所有半臂染色体的拷贝数,通过整合与健康人基线(baseline)样本的对应染色体拷贝数变化最大的五条半臂染色体的z-score,计算pascore (plasma aneuploidy score)。
16.在一些实施方案中,所述整合分类模块是svm-整合分类模块,将上述预测值k、f、s和pascore使用线性svm模型进行整合,得到最终的单一癌变可能性的预测值z。
17.在一些具体的实施方案中,所述系统中的所述文库构建装置包括:血浆游离dna提取模块,用于从血浆样品提取其中的游离dna;酶反应模块,使用酶使游离dna中的5-甲基胞嘧啶(5-mc)转化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),非甲基化胞嘧啶(c)转化为尿嘧啶(u);pcr反应模块,利用pcr对酶反应后的游离dna进行扩增。
18.在一些具体的实施方案中,所述使用的酶是tet2酶和apobec酶。
19.在一些具体的实施方案中,所述测序装置选自illumina novaseq 6000、illumina nextseq500、mgi dnbseq-t7或者mgi seq-2000。
20.在一些具体的实施方案中,所述md-knn分析模块中的md值通过以下公式计算:其中为样本n的第i个bin的md值,为第i个bin内的所有甲基化c的总数,为第i个bin内的所有c的总数。
21.在一些具体的实施方案中,所述fsi-svm分析模块中的fsi值通过以下公式计算:其中为样本n的第i个bin的fsi值,为第i个bin内的短片段数量,为第i个bin内的长片段数量。
22.在一些具体的实施方案中,所述motif-svm分析模块中的基序占比通过以下公式计算:其中为样本n的第i种4-mer基序的占比,为第i种4-mer基序的数量。
23.在一些具体的实施方案中,所述cin-pascore分析模块中的pascore通过以下公式计算:其中,为样本n的半臂染色体i相对于基线样本的z-score,为样本n的半臂染色体i的读段(reads)数,为基线样本的半臂染色体i的读段数的平均值,为基线样本的半臂染色体i的读段数的标准差;取待测样本n的z-score绝对值最大的5个半臂染色体的z-score及基线样本对应
的半臂染色体的z-score进行后续分析其中,为样本n的5个半臂染色体的z-score在自由度为3的t分布中的p值的对数和的负值;其中为样本n的pascore, 为基线样本的logp平均值,为基线样本的logp的标准差。
24.在一些具体的实施方案中,其中所述信息分析装置包括数据预处理模块,将测序装置获得的下机fastq数据转换为各模块可使用的bam文件,并建立索引。例如,进行比对、去重、排序标记、筛选并建立索引。
25.第二方面,本文还提供了基于游离dna的基因组癌变信息检测方法,其通过使用以上第一方面所述的系统进行。
26.所述基于游离dna的基因组癌变信息检测方法包括:文库构建,通过利用酶使待测样品中游离dna(例如血浆中的游离dna)中的5-甲基胞嘧啶(5-mc)转化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),非甲基化胞嘧啶(c)转化为尿嘧啶(u),用于构建文库;全基因组测序,对所构建的文库进行测序;测序信息分析,其包括以下一个或多个分析步骤:甲基化分析,用于分析游离dna的甲基化信息,片段长度系数分析,用于分析游离dna的片段化信息,末端基序分析,用于分析游离dna的片段化信息,染色体不稳定性分析,用于分析染色体的拷贝数变异信息。
27.在一些具体的实施方案中,测序信息分析还包括整合分类步骤,用于将所述甲基化分析、片段长度系数分析、末端基序分析和/或染色体不稳定性分析所获得的信息进行整合。
28.在一些具体的实施方案中,所述甲基化分析包括通过非重叠滑窗方法将人参考基因组化分为区间(例如1mb大小),计算每个区间的所有cpg位点中甲基化位点的比例,即甲基化密度md值,通过knn模型计算癌变可能性的预测值k,简称为md-knn分析。
29.在一些具体的实施方案中,所述片段长度系数分析包括通过非重叠滑窗方法将人参考基因组化分为区间(例如5mb大小),计算每个区间的短片段(例如101-167bp)和长片段(例如170-250bp)数目的比例,得到每个样本的片段长度系数fsi值,通过svm模型计算癌变可能性的预测值f,即fsi-svm分析。
30.在一些具体的实施方案中,所述末端基序分析包括计算样本的片段的5’末端4-mer基序序列的占比,通过svm模型计算癌变可能性的预测值s,即motif-svm分析。
31.在一些具体的实施方案中,所述染色体不稳定性分析包括计算样本的所有半臂染色体的拷贝数,通过整合与健康人基线样本的对应染色体拷贝数变化最大的五条半臂染色体的z-score,计算pascore值,即cin-pascore分析。
32.在一些具体的实施方案中,所述svm-整合分类包括将上述预测值k、f、s和pascore使用线性svm模型进行整合,得到最终的单一癌变可能性的预测值z,即svm-整合分类。
33.在一些具体的实施方案中,所述文库构建包括:从血浆样品提取其中的游离dna(cfdna);酶反应步骤,使用酶使游离dna中的5-甲基胞嘧啶(5-mc)转化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),非甲基化胞嘧啶(c)转化为尿嘧啶(u);和pcr扩增,利用pcr对酶反应后的游离dna进行扩增。
34.在一些具体的实施方案中,所述酶是tet2酶和apobec酶。
35.在一些具体的实施方案中,所述测序使用以下进行:illumina novaseq 6000、illumina nextseq500、mgi dnbseq-t7或者mgi seq-2000。
36.在一些具体的实施方案中,所述md-knn分析模块中的md值通过以下公式计算:其中为样本n的第i个bin的md值,为第i个bin内的所有甲基化c的总数,为第i个bin内的所有c的总数。
37.在一些具体的实施方案中,所述fsi-svm分析模块中的fsi值通过以下公式计算:其中为样本n的第i个bin的fsi值,为第i个bin内的短片段数量,为第i个bin内的长片段数量。
38.在一些具体的实施方案中,所述motif-svm分析模块中的基序占比通过以下公式计算:其中为样本n的第i种4-mer基序的占比,为第i种4-mer基序的数量。
39.在一些具体的实施方案中,所述cin-pascore分析模块中的pascore通过以下公式计算:其中,为样本n的半臂染色体i相对于基线样本的z-score,为样本n的半臂染色体i的读段数,为基线样本的半臂染色体i的读段数的平均值,为基线样本的半臂染色体i的读段数的标准差;
取待测样本n的z-score绝对值最大的5个半臂染色体的z-score及基线样本对应的半臂染色体的z-score进行以下分析其中,为样本n的5个半臂染色体的z-score在自由度为3的t分布中的p值的对数和的负值;其中为样本n的pascore, 为基线样本的logp平均值,为基线样本的logp的标准差。
40.在一些具体的实施方案中,其中所述信息分析还进一步包括数据预处理,将测序装置获得的下机fastq数据转换为各模块可使用的bam文件,并建立索引。
附图说明
41.图1. 本发明基于cfdna的低深度全基因组测序和癌变信息检测流程示意图。
42.图2. 本发明通过全基因组甲基化密度(md)的knn模型(md-knn分析模块)进行独立验证集中多个癌种预测的roc曲线。
43.图3. 本发明通过全基因组片段长度系数(fsi)的svm模型(fsi-svm分析模块)进行独立验证集中多个癌种预测的roc曲线。
44.图4. 本发明通过片段末端特征基序占比的svm模型(motif-svm分析模块)进行独立验证集中多个癌种预测的roc曲线。
45.图5. 本发明利用pascore衡量半臂染色体不稳定性(cin-pascore分析模块)进行独立验证集中多个癌种预测的roc曲线。
46.图6. 本发明最终整合分类模块进行独立验证集中多个癌种预测的roc曲线。
具体实施方式
47.如图1所示,本发明包括低深度全基因组的测序文库构建和测序,对测序数据进行多维度特征提取以及使用机器学习构建预测模型。
48.1.cfdna全基因组测序文库制备及测序原理:本发明使用了tet2酶和apobec酶实现对非甲基化胞嘧啶(c)转化为尿嘧啶(u)。具体的,首先利用tet2酶催化5-甲基胞嘧啶(5-mc)转化为5-羟甲基胞嘧啶(5-hmc),并进一步氧化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),从而保护5-mc和5-hmc在后续的apobec脱氨反应中不被作用。apobec酶将非甲基化胞嘧啶(c)脱氨转化为尿嘧啶(u),并在随后的文库扩增pcr反应中替换为胸腺嘧啶(t)。相比较传统的bisulfate化学反应,酶法转化的反应条件温和,可以最大程度的保护dna分子的完整性,因而可以用于cfdna片段特征
的分析,并可以用于低起始量dna的文库构建。
49.方案:1)从4 ml健康人或癌症患者的血清中提取cfdna,对5ng到30ng 的cfdna使用基于tet2和apobec的酶法转化,制备测序文库。
50.2)对文库进行低深度(~20g上机数据量)的2x 100 pe测序。
51.2. 甲基化密度(methylation density, md)分析原理:肿瘤发生发展过程中的甲基化状态会在基因组发生大范围的异常,本发明通过比较待测样本与健康人基线在基因组各区域的甲基化水平的相似性,可以简单灵敏的判断血浆甲基化水平是否正常,进而推测是否含有ctdna信号。分析过程中可以使用机器学习算法进行建模,进一步提升检测灵敏度。
52.方案:1)将人参考基因组按照滑窗方式划分为1mb大小的区间,对每个样本,分别计算各区间的所有cpg位点中甲基化位点的比例,即甲基化密度(md值)。
53.2)利用健康人基线和训练集中的各癌种样本的甲基化密度训练k最邻近法(k
‑ꢀ
nearest neighbor,knn)模型,利用knn模型对测试集中的待测样本进行健康人或癌症患者的分类预测。
54.3. 片段长度系数(fragment size index,fsi)分析原理:肿瘤细胞来源的cfdna的片段长度相比非肿瘤细胞具有更大的异质性。片段长度系数fsi,即整个基因组各区域的cfdna的短片段数和长片段数的比例图谱,在健康人群中高度一致,但在癌症患者中某些区域会发生变化,可能反应了癌症相关的染色质结构或其他基因组特征的异常。本发明通过比较待测样本与健康人基线的cfdna片段长度系数,可以简单灵敏的识别是否存在肿瘤来源的ctdna。通过机器学习算法进行特征识别,可以进一步提高检测灵敏度。
55.方案:1)将人参考基因组按照滑窗方式划分为5mb大小的区间,对每个样本,分别计算各区间的短片段数目和长片段数目的比例,得到每个样本的片段长度系数。
56.2)利用健康人基线和训练集中的各癌种样本的片段长度系数训练机器学习模型,选取最优模型svm(support vector machine)对测试集中的待测样本进行健康人或癌症患者的分类预测。
57.4. 片段5’末端基序分析原理:血浆cfdna片段末端的4-mer 基序序列特征具有偏好性,可能和dna内切酶例如dnase1l3的序列识别特性有关。癌症病人的相关dna内切酶可能存在异常表达,从而导致癌症病人血浆的cfdna末端序列特征发生改变,例如ccca的比例在多个癌种中显著降低。本发明通过选取256种可能的4-mer 基序中占比最高的125种基序序列,使用机器学习模型训练识别出癌症患者的血浆末端基序特征对待测样本进行判断。
58.方案:
1)计算每个样本的cfdna片段5’末端的256种可能的4-mer 基序序列的占比。选择健康人基线中占比最高的125种基序。
59.2)利用健康人基线和训练集中的各癌种样本的片段长度系数训练机器学习模型,选取最优模型svm对测试集中的待测样本进行健康人或癌症患者的分类预测。
60.5. 染色体不稳定性(chromosome instability,cin)分析原理:拷贝数变异是癌细胞最常见的遗传特征变化之一,是发生癌症基因组不稳定的普遍机制。大部分实体瘤的特征包含染色体不稳定,表现为整个染色体或部分染色体的得失。本发明通过计算半臂水平的染色体拷贝数并与健康人基线进行统计学分析,可以直接识别肿瘤来源的染色体变异,提供一种高特异性的液体活检方法。
61.方案:1)计算每个半臂染色体的读段数。
62.2)对待测样本的每个半臂读段数与基线样本进行比较并计算z-score,选取z-score绝对值最大的五条染色体半臂,将每个z-score转化为p-value并整合得到该样本的pascore(plasma aneuploidy score)以衡量该样本的染色体拷贝数异常程度。
63.6. 整合(ensemble)模型分类器(svm-整合分类模块)的构建原理:对每个样本的wms数据进行上述四个维度的分析,可以基于不同生物学机理全面衡量待测样本是否具有肿瘤信号。利用整合模型整合各维度特征的预测结果构建基于多组学分析的分类器,可以进一步提升模型的敏感度和特异性。
64.方案:利用健康人基线和训练集中的各癌种样本的上述四个维度的预测值训练机器学习模型,选取最优模型(linear svm)作为最终的整合分类器,计算最终的单一癌变可能性的预测值。
65.除前述优点以外,本发明与现有技术相比,还具有其他许多优点。
66.例如,本发明通过检测血浆低深度全基因组甲基化图谱识别异常甲基化信号,相对于常用的目标区域捕获测序方法,无需预先利用癌组织或公共数据库进行癌症差异甲基化位点的筛选及后续的血浆cfdna验证,从而大大简化了甲基化检测的实验和数据分析流程,节约了检测成本。
67.例如,本发明利用反应条件温和的酶转法进行甲基化测序,相较于重亚硫酸盐转化的方法可以最大程度的减少对dna分子的损伤。一方面,此方法适用于低起始量cfdna建库,仅需要10ml血液所提取的cfdna便可成功建库;另一方面,此方法可保留cfdna分子的原始片段特征,从而实现对同一份cfdna文库进行甲基化、片段组学、cnv等多维度特征的整合分析,提高检测的灵敏度和特异性。
68.再例如,本发明通过直接比较待测样本与健康人基线在全基因组范围的遗传和表观遗传特征的相似性,无需针对各癌种分别进行差异位点的筛选,可以实现同时对多个癌种的检测。
实施例
69.下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅仅用于说明本发明,而不应视为限定发明的范围。实施例中未注明具体技术或条件的,按照本领域的文献所描述的技术或条件或者按照产品、仪器说明书进行。所有试剂或仪器未注明生产商者,均可以市购。
70.临床队列样本信息:本试验回顾性地选取了497例无癌症史的健康人血浆以及795例不同分期的多癌种癌症患者的血浆,并随机分组为训练集和验证集。患者的癌症种类包括了乳腺癌、结直肠癌、食管癌、胃癌、肝癌、肺癌、胰腺癌。训练集包括了352例健康人及559例癌症患者(45例乳腺癌,105例结直肠癌,44例食管癌,79例胃癌,79例肝癌,110例肺癌,83例胰腺癌,14例其他),其中34.5%为早期(i或ii期)。验证集包括145例健康人和236例癌症患者(21例乳腺癌,45例结直肠癌,18例食管癌,35例胃癌,34例肝癌,47例肺癌,36例胰腺癌),其中31.8%为早期(i或ii期)。
71.一、实验流程:1. 血浆cfdna提取1.1 每位受试者10ml全血存放在康为edta采血管中,通过在4℃以1600g转速离心10min使血浆、血细胞分层。将上层血浆转移至新离心管,再次以12000rpm转速4℃离心15min取上清以去除细胞碎屑。得到约4ml血浆,-80℃冻存备用。
72.1.2血浆样本融化后,每1ml样本中加入15μl proteinase k(20mg/ml, thermoscientific cat#eo0492)和50μl sds(20%)。血浆量不足4ml,用pbs补足。
73.1.3翻转混匀,60℃孵育20min,然后冰浴5min。
74.1.4使用magmax cell-free dna isolation试剂盒(thermoscientific cat#a29319)提取cfdna。
75.1.5使用bioanalyzer 2100 (agilent technologies)检测cfdna的提取浓度和质量。
76.2. cfdna文库构建使用甲基化文库构建试剂盒nebnext enzymatic methyl-seq kit(neb,cat# e7120),以5-30ng cfdna起始量,通过tet2酶使5-甲基胞嘧啶(5-mc)转化为5-甲酰胞嘧啶(5-fc)和5-羧基胞嘧啶(5-cac),并且通过apobec酶,使非甲基化胞嘧啶(c)脱氨转化为尿嘧啶(u),然后进行扩增建库。
77.具体文库构建过程如下:2.1 内参准备取50μl cpg全甲基化的puc19 dna和50μl cpg全非甲基化的lamdba dna混匀后加入100ul打断管中,使用m220打断仪(covaris)打断。建库时,向待测cfdna加入0.001ng的puc19 dna和0.02 ng 的lambda dna。
78.2.2 cfdna样本的准备cfdna样本起始量为5-30ng,不需要打断。
79.2.3末端修复2.3.1在冰上混合以下反应体系;
试剂体积cfdna样本(5-30ng)50μlnebnext ultra ii end prep reaction buffer7μlnebnext ultra ii end prep enzyme mix3μl总体积60μl2.3.2反应体系置于pcr仪上,按下表进行末端修复反应。
80.2.4连接adaptor2.4.1在冰上操作,将以下组分加入上步的60μl反应体系中2.4.2 20℃孵育15 min。
81.2.5连接后纯化2.5.1 上一步反应结束后,取出样本,加入110μlnebnext sample purification beads,立即使用移液器吹打混匀。
82.2.5.2室温孵育5 min。
83.2.5.3 离心管置于磁力架上5 min待液体澄清,弃去上清。
84.2.5.4 加入200μl现配80%乙醇,孵育30 s后弃去。重复一次200μl 80%乙醇清洗步骤。
85.2.5.5用10μl移液器吸尽离心管底部的残留乙醇,室温干燥3-5min至乙醇完全挥发。
86.2.5.6从磁力架取下离心管,加入29 μl elution buffer (neb),震荡混匀。室温孵育1 min。
87.2.5.7短暂离心,离心管置于磁力架上3 min待液体澄清,取28 μl放进新的pcr管中。
88.2.6 5-甲基胞嘧啶和5-羟甲基胞嘧啶氧化反应使用nebnext enzymatic methyl-seq kit(neb,cat# e7120)进行以下反应操作。
89.2.6.1 tet2 reaction buffer supplement干粉加入400μl tet2 reaction buffer,充分混合。
90.2.6.2在冰上将以下组分加入上述28 μl已连接adapter的dna:
2.6.3将500 mm fe(ii) 溶液按1:1250比例稀释。往上步混匀的产物中,加入已配好的fe(ii)。
91.。
92.充分混合并在37℃孵育1h。
93.2.6.4 反应结束后移至冰上并加入1μl stop reagent。
94.试剂体积stop reagent1μl总体积51μl充分混合。
95.2.6.5 37℃孵育30 min。
96.步骤温度时间终止氧化反应37℃30min2.7氧化后纯化2.7.1 上一步反应结束后,取出样本,加入90μlnebnext sample purification beads,立即使用移液器吹打混匀。
97.2.7.2室温孵育5 min。
98.2.7.3 离心管置于磁力架上5 min待液体澄清,弃去上清。
99.2.7.4 加入200μl现配80%乙醇,孵育30 s后弃去。重复一次200μl 80%乙醇清洗步骤。
100.2.7.5用10μl移液器吸尽离心管底部的残留乙醇,室温干燥3-5min至乙醇完全挥发。
101.2.7.6从磁力架取下离心管,加入17 μl elution buffer,震荡混匀。室温孵育1 min。
102.2.7.7短暂离心,离心管置于磁力架上3 min待液体澄清,取16 μl放进新的pcr管中。
103.2.8 dna变性2.8.1 配制新鲜的0.1n naoh。
104.2.8.2 提前预热pcr仪到50℃。
105.2.8.3加入4μl 0.1n naoh到上步16μl纯化产物中,充分混合。
106.2.8.4 50℃孵育10 min。
107.2.8.5 反应结束后立刻放入冰上。
108.2.9 胞嘧啶脱氨基2.9.1在冰上将下列组分加入上步20μl变性dna。
109.试剂体积nuclease-free water68μlapobec reaction buffer10μlbsa1μlapobec1μl总体积80μl充分混合。
110.2.9.2 在pcr仪上37℃孵育3h后转为4℃终止反应。
111.2.10脱氨后纯化2.10.1 上一步反应结束后,取出样本,加入100μlnebnext sample purification beads,立即使用移液器吹打混匀。
112.2.10.2室温孵育5 min。
113.2.10.3 离心管置于磁力架上5 min待液体澄清,弃去上清。
114.2.10.4 加入200μl 现配80%乙醇,孵育30 s后弃去。重复一次200μl 80%乙醇清洗步骤。
115.2.10.5用10μl移液器吸尽离心管底部的残留乙醇,室温干燥3-5min至乙醇完全挥发。
116.2.10.6从磁力架取下离心管,加入21 μl elution buffer,震荡混匀。室温孵育1 min。
117.2.10.7短暂离心,离心管置于磁力架上3 min待液体澄清,取20 μl放进新的pcr管中。
118.2.11文库pcr扩增2.11.1 在冰上将下列组分加入上步脱氨后的20μl dna。
119.2.11.2 充分混合后在pcr以上进行以下pcr反应。
120.2.12 pcr后纯化2.12.1 上一步反应结束后,取出样本,加入45 μlnebnext sample purification beads,立即使用移液器吹打混匀。
121.2.12.2室温孵育5 min。
122.2.12.3 离心管置于磁力架上5 min待液体澄清,弃去上清。
123.2.12.4 加入200μl 现配80%乙醇,孵育30 s后弃去。重复一次200μl 80%乙醇清洗步骤。
124.2.12.5用10μl移液器吸尽离心管底部的残留乙醇,室温干燥3-5min至乙醇完全挥发。
125.2.12.6从磁力架取下离心管,加入21 μl elution buffer,震荡混匀。室温孵育1 min。
126.2.12.7短暂离心,离心管置于磁力架上3 min待液体澄清,取20 μl放进新的pcr管中。
127.2.13 文库定量使用qubit高灵敏试剂(thermoscientific cat#q32854)对所构建的文库进行定量,文库产量大于400ng进行后续上机测序。
128.3.文库测序取100ng上述文库加入10% phix dna(illumina cat#fc-110-3001)混合成上机样品,在novaseq 6000 (illumina)平台进行pe100测序。 二、生信分析流程:1. 处理下机fastq数据为各模块可使用的bam文件1.1 去接头调用trimmomatic-0.36将每一对fastq文件都作为配对的读段(paired reads)比对到hg19人类参考基因组序列,除m参数与指定reads group的id外,不使用其余参数选项,生成初始bam文件。
129.1.2 比对调用bismark-v0.19.0将去接头后的每一对fastq文件都作为配对读段比对到hg19人类参考基因组序列和lambda dna参考基因组序列,生成初始bam文件。
130.1.3 去重
调用bismark
‑ꢀ
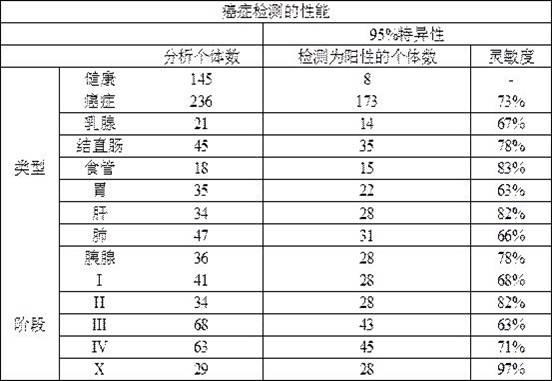
v0.19.0的deduplicate模块,对初始bam文件进行去重复处理,生成去重后的bam文件。
131.1.4 排序标记调用samtools-1.3的sort模块,对去重后的bam文件进行排序,生成排序后的bam文件。然后,调用picard-2.1.0的addorreplacereadgroups模块,对排序后的bam文件进行标记分组。
132.1.5 筛选调用bamutil-1.0.14的clipoverlap模块对标记分组后的bam文件进行筛选,去除重叠的配对读段,生成bam文件。并调用samtools-1.3 view对去除重叠的bam文件的比对质量进行过滤,采用
“‑
q 20”作为参数,生成最终bam文件。
133.1.6 建立索引调用samtools-1.3的index模块对最终生成的bam文件建立索引,生成与最终bam文件配对的bai文件。
134.2. 甲基化密度(methylation density, md)分析(md-knn分析模块)2.1将人参考基因组按照非重叠滑窗方式划分为1mb大小的区间(bin),剔除比对率差的区间后剩余1846个bin,对每个样本,分别计算这1846个bin的所有cpg位点中甲基化位点的比例,该值对应于每个样本的甲基化密度(md)值,具体公式如下:其中为样本n的第i个bin的md值,为第i个bin内的所有甲基化c的总数,为第i个bin内的所有c的总数。
135.2.2对上述2.1中获得的每个样本的1846个md值进行标准化处理计算z-score,应用r语言的philentropy包计算样本间的欧式距离(distance),样本的权重选择1/distance。用50轮模拟调整参数k,每轮用80%的训练集样本,计算k在不同取值时,根据50轮里每一轮out-of-bag(oob)的20%样本的预测结果计算auc,选择oob样本auc最高的k值。
136.2.3 用训练好的knn(k-nearest neighbor,knn)模型对测试集中的每个待测样本进行健康人或癌症患者的分类预测,获得预测值k。如图2所示,md-knn分类器对测试集中的单一癌种的检测roc曲线面积(auc)达到0.789-0.870,对全部七个癌种的检测auc性能达到0.830,显示出良好的癌症检测性能。
137.3. 片段长度系数(fragment size index,fsi)分析(fsi-svm分析模块)3.1将人参考基因组按照非重叠滑窗方式划分为5mb大小的区间(bin),剔除比对率差的黑名单区间后剩余502个bin,分别计算这502个bin内的短片段(101-167bp)数目和长片段(170-250bp)数目的比例,并用loess算法进行gc矫正,得到每个样本的片段长度系数(fsi)。具体计算公式如下:其中为样本n的第i个bin的fsi值,为第i个bin内的短片段数量,为第i个bin内的长片段数量。
138.3.2对每个样本的502个fsi值应用python的sklearn包训练svm(support vector machine, svm)模型,使用网格搜索的方式进行超参数的选择,进行10乘交叉验证获得超参数。
139.3.3对测试集中的每个待测样本进行健康人或癌症患者的分类预测,获得预测值f。如图3所示,fsi-svm分类器对测试集中的单一癌种的检测roc曲线面积(auc)达到0.874-0.933,对全部七个癌种的检测auc性能达到0.904,显示出良好的癌症检测性能。
140.4. 片段末端基序分析(motif-svm分析模块)4.1 计算每个样本的片段5’末端的256种(即四种碱基可能的排列组合,4的4次方)可能的4-mer 基序序列的占比。选择占比超过0.0004且在健康人基线中占比最高的125种基序,如下表1所示。
141.表1上述基序占比通过以下公式计算:其中为样本n的第i种4-mer基序的占比,为第i种4-mer基序的数
量。
142.4.2利用健康人基线和训练集中的所有癌症样本的125种特征基序的占比,应用r语言的caret包训练svm模型,使用网格搜索的方式进行超参数的选择,进行10乘交叉验证。
143.4.3 对测试集中的每个待测样本进行健康人或癌症患者的分类预测,获得预测值s。如图4所示,motif-svm分类器对测试集中的单一癌种的检测roc曲线面积(auc)达到0.920-0.966,对全部七个癌种的检测auc性能达到0.943,显示出良好的癌症检测性能。
144.5. 染色体不稳定性(chromosome instability,cin)分析(cin-pascore分析模块)5.1 对每个样本,计算每半臂染色体的loess算法gc矫正后的读段数。
145.5.2 以训练集中的352例健康人作为基线样本,对待测样本的每半臂染色体读段数对应基线样本的相应半臂染色体读段数的均值和标准差进行z-score转化。
146.5.3 待测样本选择z-score绝对值最大的5条半臂染色体及基线样本对应的半臂染色体的z-score按文献所述方式(leary et al., 2012 sci transl med,)计算pascore。具体计算如下。
147.其中,为样本n的半臂染色体i相对于基线样本的z-score,为样本n的半臂染色体i的读段数,为基线样本的半臂染色体i的读段数的平均值,为基线样本的半臂染色体i的读段数的标准差;取待测样本n的z-score绝对值最大的5个半臂染色体的z-score及基线样本对应的半臂染色体的z-score进行后续分析其中,为样本n的5个半臂染色体的z-score在自由度为3的t分布中的p值的对数和的负值;其中为样本n的pascore, 为基线样本的logp平均值,为基线样本的logp的标准差。
148.5.4 如图5所示,cin-pascore算法对测试集中单一癌种检测的auc达到0.770-0.854,对全部七个癌种的检测auc性能达到0.812。
149.6. 整合模型分类器的构建(svm-整合分类模块)6.1 将上述所得每个样本的md-knn、fsi-svm、motif-svm、cin-pascore数值(即上述预测值k、f、s和pascore)作为训练模型中的特征。
150.6.2 应用r语言的caret包训练linearsvm模型,使用网格搜索的方式进行超参数
的选择,进行10乘交叉验证。通过训练好的模型对测试集中的每个样本进行预测,获得样本预测为癌症单一癌变可能性的预测值z。
151.6.3 如图6所示,本发明的整合模型分类器对测试集中单一癌种检测的auc达到0.934
–
0.971,对全部七个癌种的检测auc达到0.952,性能超过任何单一的遗传或表观遗传特征分类器,展示出了多维度整合分析癌变信息数据相对单一组学的优越性。
152.6.4 如表2所示,本发明的整合模型分类器在95%特异性下对测试集中七个癌种的检测灵敏度均在60%以上,对于早期癌症(i或ii期)的检测灵敏度可达75%,展示出了对于各癌种良好的检测性能,并具有极大的潜力应用于癌症早期筛查。
153.表2. 本发明的整个分类模块在95%特异性下对验证集中各癌种及各分期的检测灵敏度。
154.。