1.本发明属于有机合成技术领域,具体涉及一种邻位环己二酮的制备方法。
背景技术:
2.邻位环己二酮是一种重要的医药合成中间体,可用于合成多种杂环化合物。此外,邻位环己二酮也是一种具有焦甜香气的香料,常被用来制备香料,因此市场需求量较大。
3.目前,工业化生产邻位环己二酮大多采用亚硒酸氧化环己酮制得,该方法先将环己酮加热至70~80℃,然后逐滴加入亚硒酸与乙醇的混合溶液,滴加完毕进行加热回流2h,最后进行干燥分离,提纯得到邻位环己二酮,收率在30~50%。现有技术中所公开的制备方法所得到的邻位环己二酮的产率都较低。
技术实现要素:
4.本发明的目的在于提供一种邻位环己二酮的制备方法,本发明所提供的制备方法,邻位环己二酮的产率较高。
5.为了实现上述目的,本发明提供如下技术方案:
6.本发明提供了一种邻位环己二酮的制备方法,包括以下步骤:
7.将环己酮、二元醇、酸性催化剂和极性溶剂ⅰ经第一混合后,进行缩合反应,得到化合物ⅰ;所述二元醇为乙二醇或丙二醇;
8.将所述化合物ⅰ、溴代试剂和极性溶剂ⅱ经第二混合后,进行溴代反应,得到化合物ⅱ;
9.将所述化合物ⅱ、酸性试剂和极性溶剂ⅲ经第三混合后,进行水解反应ⅰ,得到含有化合物ⅲ的反应液;
10.将所述含有化合物ⅲ的反应液和碱性试剂经第四混合后,进行水解反应ⅱ,得到所述邻位环己二酮;
11.所述化合物ⅰ、化合物ⅱ和化合物ⅲ具有如下所示结构:
[0012][0013]
其中r为-ch
2-ch
2-或-ch
2-ch
2-ch
2-。
[0014]
优选的,所述酸性催化剂包括浓硫酸、对甲苯磺酸、氯化铝和强酸性阳离子交换树脂中的一种或几种;
[0015]
所述溴代试剂包括n-溴代琥珀酰亚胺、苯基三甲基三溴化铵、二溴异氰尿酸、液溴、n-溴乙酰胺、n-溴丁二酰亚胺和苄基三溴铵盐中的一种或几种。
[0016]
优选的,所述环己酮和二元醇的摩尔比为1:(3~5);
[0017]
所述酸性催化剂的质量为环己酮质量的5%~20%;
[0018]
所述环己酮和极性溶剂ⅰ的用量比为1g:(5~10)ml。
[0019]
优选的,所述缩合反应的温度为60~100℃,时间为1~3h。
[0020]
优选的,所述化合物ⅰ和溴代试剂的摩尔比为1:(0.2~2.0);
[0021]
所述化合物ⅰ和极性有机溶剂ⅱ的用量比为1g:(5~15)ml。
[0022]
优选的,所述溴代反应的温度为25~40℃,时间为1~5h。
[0023]
优选的,所述酸性试剂的质量浓度为10%~30%;
[0024]
所述碱性试剂的质量浓度为10%~30%。
[0025]
优选的,所述化合物ⅱ和酸性试剂的用量比为1g:(3~6)ml;
[0026]
所述化合物ⅱ和极性试剂ⅲ的用量比为1g:(2~10)ml。
[0027]
优选的,所述水解反应ⅰ的温度为25~40℃,时间为1~2h。
[0028]
优选的,所述水解反应ⅱ的温度为25~40℃,时间为2~4h。
[0029]
本发明提供了一种邻位环己二酮的制备方法,包括以下步骤:将环己酮、二元醇、酸性催化剂和极性溶剂ⅰ经第一混合后,进行缩合反应,得到化合物ⅰ;将所述化合物ⅰ、溴代试剂和极性溶剂ⅱ经第二混合后,进行溴代反应,得到化合物ⅱ;将所述化合物ⅱ、酸性试剂和极性溶剂ⅲ经第三混合后,进行水解反应ⅰ,得到含有化合物ⅲ的反应液;将所述含有化合物ⅲ的反应液和碱性试剂经第四混合后,进行水解反应ⅱ,得到所述邻位环己二酮。本发明利用环己酮作为原料,经缩合、溴代和水解反应得到邻位环己二酮,产率高。
附图说明
[0030]
图1为实施例1得到的化合物ⅰ的气相色谱图;
[0031]
图2为实施例1得到的化合物ⅱ的气相色谱图;
[0032]
图3为实施例1得到的化合物ⅲ的气相色谱图;
[0033]
图4为实施例1得到的邻位环己二酮的气相色谱图;
[0034]
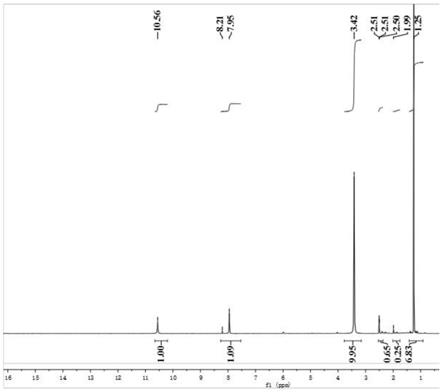
图5为实施例1得到的邻位环己二酮的核磁氢谱图。
具体实施方式
[0035]
本发明提供了一种邻位环己二酮的制备方法,包括以下步骤:
[0036]
将环己酮、二元醇、酸性催化剂和极性溶剂ⅰ经第一混合后,进行缩合反应,得到化合物ⅰ;所述二元醇为乙二醇或丙二醇;
[0037]
将所述化合物ⅰ、溴代试剂和极性溶剂ⅱ经第二混合后,进行溴代反应,得到化合物ⅱ;
[0038]
将所述化合物ⅱ、酸性试剂和极性溶剂ⅲ经第三混合后,进行水解反应ⅰ,得到含有化合物ⅲ的反应液;
[0039]
将得到的含有化合物ⅲ的反应液和碱性试剂经第四混合后,进行水解反应ⅱ,得到所述邻位环己二酮;
[0040]
所述化合物ⅰ、化合物ⅱ和化合物ⅲ具有如下所示结构:
[0041][0042]
其中r为-ch
2-ch
2-或-ch
2-ch
2-ch
2-。
[0043]
在本发明中,若无特殊说明,所有制备原料均为本领域技术人员熟知的市售产品。
[0044]
本发明的合成路线优选为:
[0045][0046]
本发明将环己酮、二元醇、酸性催化剂和极性溶剂ⅰ经第一混合后,进行缩合反应,得到化合物ⅰ;所述二元醇为乙二醇或丙二醇。
[0047]
在本发明中,所述酸性催化剂优选包括浓硫酸、对甲苯磺酸、氯化铝和强酸性阳离子交换树脂中的一种或几种;当所述酸性催化剂为上述具体选择中的两种以上时,本发明对所述物质的比例没有特殊的限定,按照任意比例混合均可。在本发明中,所述极性溶剂ⅰ优选包括甲苯、苯、二氧六环、n,n-二甲基甲酰胺、二甲亚砜和n-甲基吡咯烷酮中的一种或几种;当所述极性溶剂ⅰ为上述具体选择中的两种以上时,本发明对所述物质的比例没有特殊的限定,按照任意比例混合均可。
[0048]
在本发明中,所述环己酮和二元醇的摩尔比优选为1:(3~5),进一步优选为1:4。在本发明中,所述酸性催化剂的质量优选为环己酮质量的5%~20%,进一步优选为8%~18%,更优选为10%~15%。在本发明中,所述环己酮和极性溶剂ⅰ的用量比优选为1g:(5~10)ml,进一步优选为1g:(6~9)ml,更优选为1g:(7~8)ml。
[0049]
在本发明中,所述第一混合的过程优选在搅拌的条件下进行。在本发明中,所述搅拌的转速优选为500~1000rpm,进一步优选为600~900rpm,更优选为700~800rpm;时间优选为30~60min,进一步优选为35~55min,更优选为40~50min。在本发明中,所述搅拌的方式优选为磁力搅拌。
[0050]
在本发明中,所述缩合反应的温度优选为60~100℃,进一步优选为70~90℃,更优选为75~85℃;时间优选为1~3h,进一步优选为1.2~2.8h,更优选为1.5~2.5h。在本发明中,所述缩合反应优选采用回流的方式进行。本发明对所述回流的过程没有特殊的限定,采用本领域技术人员熟知的即可。
[0051]
所述缩合反应完成后,本发明优选对反应产物进行后处理;所述后处理优选包括依次进行的冷却、ph值调节、淬灭、分离、萃取、干燥和旋蒸。
[0052]
本发明对所述冷却的过程没有特殊的限定,采用本领域技术人员熟知的即可。在本发明中,所述冷却后的温度优选为30~40℃,进一步优选为32~38℃,更优选为35~36℃。
[0053]
在本发明中,所述ph值调节所采用的的调节试剂优选为碳酸钠和/或碳酸钾。本发明对所述调节试剂的用量没有特殊的限定,只要能够将产物的ph值调节到中性即可。
[0054]
在本发明中,所述淬灭所采用的淬灭剂优选为水。在本发明中,所述淬灭的过程优选为将ph值调节后的产物和水混合进行淬灭。在本发明中,所述ph值调节后的产物和水的体积比优选为1:(1~2)。
[0055]
在本发明中,所述分离的过程优选为将淬灭得到的产物进行分离,得到有机相和水相。本发明对所述分离的方式没有特殊的要求,采用本领域技术人员熟知的即可。
[0056]
所述分离完成后,本发明还优选包括将得到的有机相进行水洗。在本发明中,所述有机相和水的体积比优选为1:(1~2)。在本发明中,所述洗涤的次数的优选为3次。
[0057]
所述水洗完成后,本发明还优选包括将水洗得到的水相和所述分离得到的水相进行混合,得到混合水相。
[0058]
在本发明中,所述萃取所采用的萃取剂优选为乙酸乙酯和/或石油醚。在本发明中,所述萃取的过程优选为将所述混合水相和萃取剂混合进行萃取。在本发明中,所述混合水相和萃取剂的体积比优选为1:(1~2)。
[0059]
所述萃取完成后,本发明优选对萃取得到的有机相进行干燥,所述干燥的过程优选为将有机相和干燥剂混合进行干燥。在本发明中,所述干燥剂优选为无水硫酸钠。本发明对所述干燥剂的用量没有特殊的限定,只要能够将所述有机相中的多余水分去除即可。
[0060]
本发明对所述旋蒸的过程没有特殊的限定,采用本领域技术人员熟知的即可。
[0061]
在本发明中,所述化合物ⅰ优选具有如下所示结构:进一步优选为
[0062]
在本发明中,所述化合物ⅰ的产率优选为81.9%~88%;纯度优选为96%~98.9%。在本发明中,利用本发明提供的酸性催化剂催化环己酮和二元醇的缩合反应,能够进一步提高化合物ⅰ的产率和纯度。
[0063]
得到所述化合物ⅰ后,本发明将所述化合物ⅰ、溴代试剂和极性溶剂ⅱ经第二混合后,进行溴代反应,得到化合物ⅱ。
[0064]
在本发明中,所述溴代试剂优选包括n-溴代琥珀酰亚胺、苯基三甲基三溴化铵、二溴异氰尿酸、液溴、n-溴乙酰胺、n-溴丁二酰亚胺和苄基三溴铵盐中的一种或几种;当所述溴代试剂为上述具体选择中的两种以上时,本发明对所述物质的比例没有特殊的要求,按照任意比例混合均可。
[0065]
在本发明中,所述极性溶剂ⅱ优选包括三氯甲烷、二氯甲烷、乙腈、乙醚、四氢呋喃和石油醚中的一种或几种;当所述极性溶剂ⅱ为上述具体选择中的两种以上时,本发明对所述物质的比例没有特殊的要求,按照任意比例混合均可。
[0066]
在本发明中,所述化合物ⅰ和溴代试剂的摩尔比优选为1:(0.2~2.0),进一步优选为1:(0.4~1.8),更优选为1:(0.6~1.6)。在本发明中,所述化合物ⅰ和极性溶剂ⅱ的用量比优选为1g:(5~15)ml,进一步优选为1g:(6~14)ml,更优选为1g:(7~13)ml。
[0067]
在本发明中,所述第二混合优选包括以下步骤:将化合物ⅰ和极性溶剂ⅱ预混后得到预混液;将所述预混液和溴代试剂进行再混合。
[0068]
在本发明中,所述预混优选在搅拌的条件下进行。在本发明中,所述搅拌的转速优选为400~1000rpm,进一步优选为500~900rpm,更优选为600~800rpm。在本发明中,所述再混合的方式优选将所述溴代试剂滴加到所述预混液中。在本发明中,所述滴加的速度优选为30ml/min。
[0069]
在本发明中,所述溴代反应的温度优选为25~40℃,进一步优选为28~38℃,更优选为30~35℃;时间为1~5h,进一步优选为2~4h,更优选为3h。在上述条件下进行溴代反应,能够进一步提高化合物ⅱ的产率和纯度
[0070]
所述溴代反应完成后,本发明还优选包括对反应产物进行后处理;所述后处理优选包括依次进行的洗涤、萃取和旋蒸。
[0071]
在本发明中,所述洗涤的过程优选为:将反应产物和碳酸氢钠溶液混合后,进行洗涤。在本发明中,所述碳酸氢钠溶液的质量浓度优选为5%~20%,进一步优选为8%~18%,进一步优选为10%~15%。在本发明中,所述反应产物和碳酸氢钠溶液的体积比优选为(1~2):1。在本发明中,所述洗涤的次数优选为3次。
[0072]
在本发明中,所述萃取所采用的萃取剂优选为乙酸乙酯和/或石油醚。本发明对所述萃取剂的用量和萃取的过程没有特殊的要求,只要能够将产物全部萃取出即可。
[0073]
本发明对所述旋蒸的过程没有特殊的限定,采用本领域技术人员熟知的即可。
[0074]
在本发明中,所述化合物ⅱ优选具有如下所示结构:进一步优选为
[0075]
在本发明中,所述化合物ⅱ的产率优选为85%~89%;纯度优选为96%~98.7%。
[0076]
得到所述化合物ⅱ后,本发明将所述化合物ⅱ、酸性试剂和极性溶剂ⅲ经第三混合后,进行水解反应ⅰ,得到含有化合物ⅲ的反应液。
[0077]
在本发明中,所述酸性试剂优选包括盐酸、硫酸、碳酸或醋酸中的一种或几种;当所述酸性试剂为上述具体选择中的两种以上时,本发明对所述所述物质的比例没有特殊的限定,按照任意比例混合均可。在本发明中,所述酸性试剂的质量浓度优选为10%~30%,进一步优选为12%~28%,更优选为15%~25%。
[0078]
在本发明中,所述极性溶剂ⅲ优选包括甲醇溶液和/或乙醇溶液;当所述极性溶剂ⅲ为甲醇溶液和乙醇溶液时,本发明对两者的比例没有特殊的限定,按照任意比例混合均可。在本发明中,所述极性溶剂ⅲ的质量浓度优选为60%~80%,进一步优选为65%~75%,更优选为68%~70%。
[0079]
在本发明中,所述化合物ⅱ和酸性试剂的用量比为1g:(3~6)ml,进一步优选为1g:(3.5~5.5)ml,更优选为1g:(4~5)ml。在本发明中,所述化合物ⅱ和极性试剂ⅲ的用量比为1g:(2~10)ml,进一步优选为1g:(3~9)ml,更优选为1g:(4~8)ml。
[0080]
在本发明中,所述第三混合优选在搅拌的条件下进行。在本发明中,所述搅拌的转速优选为500~1000rpm,进一步优选为600~900rpm,更优选为700~800rpm;时间优选为1~2h。
[0081]
在本发明中,所述水解反应ⅰ的温度优选为25~40℃,进一步优选为28~38℃,更优选为30~35℃;时间优选为1~2h,进一步优选为1.2~1.8h,更优选为1.4~1.6h。所述水解反应ⅰ优选通过tlc监测反应是否完成。
[0082]
在本发明中,所述化合物ⅲ的纯度优选为94.5%~98.6%。
[0083]
得到所述含有化合物ⅲ的反应液后,本发明将所述含有化合物ⅲ的反应液和碱性试剂经第四混合后,进行水解反应ⅱ,得到所述邻位环己二酮。
[0084]
在本发明中,所述碱性试剂优选包括n,n-二异丙基乙胺、叔丁醇钾、醋酸钾、磷酸钾、三乙胺、碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、三乙胺和吡啶中的一种或几种;当所述碱性试剂为上述具体选择中的两种以上时,本发明对所述物质的比例没有特殊的限定,按照任意比例混合均可。在本发明中,所述碱性试剂的质量浓度优选为10%~30%,进一步优选为12%~28%,更优选为15%~25%。
[0085]
本发明对所述碱性试剂的用量没有特殊要求,只要能够将混合液体的ph值调整为中性即可。
[0086]
在本发明中,所述第四混合优选在搅拌的条件下进行;所述搅拌的转速优选为1000rpm;时间优选为1~2h,进一步优选为1.5h。
[0087]
在本发明中,所述水解反应ⅱ的温度优选为25~40℃,进一步优选为28~38℃,更优选为30~35℃;时间优选为2~4h,进一步优选为1.2~1.8h,更优选为1.4~1.6h。在本发明中,所述水解反应ⅱ优选通过tlc监测反应是否完成。
[0088]
所述水解反应ⅱ完成后,本发明优选对反应产物进行后处理;所述后处理优选包括依次进行的干燥、旋蒸和重结晶。
[0089]
在本发明中,所述干燥的过程优选为将反应产物和无水硫酸钠混合,进行干燥。本发明对所述无水硫酸钠的用量没有特殊的要求,能够将反应产物中的多余水分去除即可。
[0090]
本发明对所述旋蒸的过程没有特殊的限定,采用本领域技术人员熟知的即可。
[0091]
在本发明中,所述重结晶采用的重结晶试剂优选为甲醇和/或乙醇。在本发明中,所述旋蒸得到的粗产品和所述重结晶试剂的用量比优选为1g:(0.5~1)ml,进一步优选为1g:(0.6~0.9)ml。
[0092]
在本发明中,所述邻位环己二酮的产率优选为90.6%~97%;纯度优选为96%~99.2%。
[0093]
在本发明中,所述邻位环己二酮的总产率优选为65.7%~76%。
[0094]
本发明所采用的原料廉价易得,采用的试剂均为低毒性,安全且环保。
[0095]
本发明的制备方法操作简单,反应条件温和,不需要高温反应,能耗较低,经济效益高;且反应过程中的副产物少,易分离,适合工业化生产。
[0096]
为了进一步说明本发明,下面结合附图和实施例对本发明提供的一种邻位环己二酮的制备方法进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
[0097]
实施例1
[0098]
取4.38g环己酮、10g乙二醇、0.4g对甲苯磺酸和50ml甲苯,在磁力搅拌器下以
500rpm的转速搅拌30min使反应液混合均匀,在80℃下回流1h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入1g碳酸钠,搅拌1h,然后倒入100ml水中进行淬灭后,分离得到有机相和水相;将有机相用20ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入50ml乙酸乙酯进行提取,然后向乙酸乙酯中加入3g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为82.6%,纯度为98.3%。
[0099]
在25℃下,将3g化合物ⅰ和20ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加1ml液溴,滴加完毕继续搅拌反应1h。tlc监测反应进程,反应结束向反应液中加入10ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用20ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为87.1%,纯度为97.7%。
[0100]
将1.4g化合物ⅱ、10ml质量浓度为60%的甲醇溶液和5ml质量浓度为10%的硫酸混合,在温度为25℃、转速为500rpm下搅拌反应1h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为98%);将含有化合物ⅲ的反应液和10ml质量浓度20%的氢氧化钠溶液混合,将ph值调节为中性,在35℃下以1000rpm的搅拌速度搅拌反应1h,tlc监测反应进程,反应结束后,向反应液中加入1g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和10ml乙醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为96%,纯度为98.9%;总产率为69.1%。
[0101]
实施例2
[0102]
取98g环己酮、200g乙二醇、8g对甲苯磺酸和1000ml甲苯,在磁力搅拌器下以500rpm的转速搅拌60min使反应液混合均匀,在80℃下回流3h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入20g碳酸钠,搅拌2h,然后倒入2000ml水中进行淬灭后,分离得到有机相和水相;将有机相用400ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入200ml乙酸乙酯进行提取,然后向乙酸乙酯中加入60g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为86%,纯度为96.4%。
[0103]
在25℃下,将60g化合物ⅰ和400ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加20ml液溴,滴加完毕继续搅拌反应3h。tlc监测反应进程,反应结束向反应液中加入200ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用400ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为88%,纯度为96%。
[0104]
将30g化合物ⅱ、200ml质量浓度为60%的甲醇溶液和100ml质量浓度为10%的硫酸混合,在温度为30℃、转速为500rpm下搅拌反应2h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为98%);将含有化合物ⅲ的反应液和200ml质量浓度20%的氢氧化钠溶液混合,将ph值调节为中性,在35℃下以1000rpm的搅拌速度搅拌反应3h,tlc监测反应进程,反应结束后,向反应液中加入10g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和100ml乙醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为95.9%,,纯度为98.3%;总产率为72.6%。
[0105]
实施例3
[0106]
取9.8g环己酮、20g乙二醇、1.0g浓硫酸和100ml甲苯,在磁力搅拌器下以600rpm的转速搅拌10min使反应液混合均匀,在70℃下回流2h进行缩合反应;反应完成后,将反应产
物冷却至30℃;向反应产物加入2g碳酸钾,搅拌2h,然后倒入200ml水中进行淬灭后,分离得到有机相和水相;将有机相用40ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入100ml石油醚进行提取,然后向石油醚中加入6g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为85.2%,纯度为98.2%。
[0107]
在30℃下,将5g化合物ⅰ和20ml三氯甲烷混合后,在转速为600rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加1mln-溴代琥珀酰亚胺,滴加完毕继续搅拌反应1h。tlc监测反应进程,反应结束向反应液中加入20ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用40ml石油醚萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为87.1%,纯度为97.3%。
[0108]
将3g化合物ⅱ、10ml质量浓度为60%的乙醇溶液和10ml质量浓度为10%的盐酸混合,在温度为25℃、转速为600rpm下搅拌反应1h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为94.5%);将含有化合物ⅲ的反应液和10ml质量浓度20%的氢氧化钾溶液混合,将ph值调节为中性,在35℃下以1000rpm的搅拌速度搅拌反应1h,tlc监测反应进程,反应结束后,向反应液中加入1g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和20ml甲醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为91.6%,纯度为99.2%;总产率为68%。
[0109]
实施例4
[0110]
取19.5g环己酮、40g乙二醇、1.5g氯化铝和200ml二氧六环,在磁力搅拌器下以400rpm的转速搅拌20min使反应液混合均匀,在90℃下回流1.5h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入4g碳酸钾,搅拌1h,然后倒入400ml水中进行淬灭后,分离得到有机相和水相;将有机相用20ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入200ml乙酸乙酯进行提取,然后向乙酸乙酯中加入g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为84%,纯度为97.9%。
[0111]
在35℃下,将10g化合物ⅰ和70ml乙醚混合后,在转速为400rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加5ml四丁基溴化铵,滴加完毕继续搅拌反应2h。tlc监测反应进程,反应结束向反应液中加入70ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用70ml石油醚萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为85%,纯度为97%。
[0112]
将5g化合物ⅱ、10ml质量浓度为60%的甲醇溶液和20ml质量浓度为30%的硫酸混合,在温度为35℃、转速为400rpm下搅拌反应1h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为93%);将含有化合物ⅲ的反应液和40ml质量浓度30%的碳酸钾溶液混合,将ph值调节为中性,在35℃下以1000rpm的搅拌速度搅拌反应3h,tlc监测反应进程,反应结束后,向反应液中加入4g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和40ml甲醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为92%,纯度为97.8%;总产率为65.7%。
[0113]
实施例5
[0114]
取50g环己酮、100g乙二醇、4g对甲苯磺酸和500mln,n-二甲基甲酰胺,在磁力搅拌器下以500rpm的转速搅拌60min使反应液混合均匀,在100℃下回流1h进行缩合反应;反应完成后,将反应产物冷却至40℃;向反应产物加入10g碳酸钠,搅拌2h,然后倒入500ml水中进行淬灭后,分离得到有机相和水相;将有机相用200ml水进行洗涤,洗涤三次,将洗涤得到
的水相和分离得到的水相进行混合后,加入100ml乙酸乙酯进行提取,然后向乙酸乙酯中加入30g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为88%,纯度为98.9%。
[0115]
在40℃下,将30g化合物ⅰ和200ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加10ml液溴,滴加完毕继续搅拌反应1h。tlc监测反应进程,反应结束向反应液中加入100ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用200ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为89%,纯度为98%。
[0116]
将15g化合物ⅱ、100ml质量浓度为60%的甲醇溶液和50ml质量浓度为10%的硫酸混合,在温度为40℃、转速为500rpm下搅拌反应1h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为98.6%);将含有化合物ⅲ的反应液和100ml质量浓度20%的碳酸钠溶液混合,将ph值调节为中性,在40℃下以1000rpm的搅拌速度搅拌反应2h,tlc监测反应进程,反应结束后,向反应液中加入5g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和50ml乙醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为97%,纯度为98.1%;总产率为76%。
[0117]
实施例6
[0118]
取5g环己酮、10g乙二醇、0.4g酸性阳离子交换树脂和50ml甲苯,在磁力搅拌器下以500rpm的转速搅拌30min使反应液混合均匀,在80℃下回流1h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入1g碳酸钠,搅拌1h,然后倒入100ml水中进行淬灭后,分离得到有机相和水相;将有机相用20ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入50ml乙酸乙酯进行提取,然后向乙酸乙酯中加入3g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为81.9%,纯度为98.7%。
[0119]
在25℃下,将3g化合物ⅰ和10ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加2ml液溴,滴加完毕继续搅拌反应2h。tlc监测反应进程,反应结束向反应液中加入10ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用10ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为88.1%,纯度为98.7%。
[0120]
将2g化合物ⅱ、15ml质量浓度为80%的甲醇溶液和5ml质量浓度为20%的硫酸混合,在温度为28℃、转速为500rpm下搅拌反应1h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为98.3%);将含有化合物ⅲ的反应液和15ml质量浓度10%的氢氧化钠溶液混合,将ph值调节为中性,在40℃下以1000rpm的搅拌速度搅拌反应1h,tlc监测反应进程,反应结束后,向反应液中加入1g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和15ml乙醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为93%,纯度为96.9%;总产率为67.1%。
[0121]
实施例7
[0122]
取98g环己酮、200g乙二醇、10g浓硫酸和1000ml苯,在磁力搅拌器下以1000rpm的转速搅拌30min使反应液混合均匀,在85℃下回流2h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入20g碳酸钾,搅拌3h,然后倒入1000ml水中进行淬灭后,分离得到有机相和水相;将有机相用400ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得
到的水相进行混合后,加入1000ml石油醚进行提取,然后向乙酸乙酯中加入60g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为87%,纯度为96%。
[0123]
在35℃下,将50g化合物ⅰ和200ml三氯甲烷混合后,在转速为1000rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加30mln-溴代琥珀酰亚胺,滴加完毕继续搅拌反应2h。tlc监测反应进程,反应结束向反应液中加入200ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用400ml石油醚萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为86.1%,纯度为97.1%。
[0124]
将30g化合物ⅱ、100ml质量浓度为60%的乙醇溶液和100ml质量浓度为10%的盐酸混合,在温度为38℃、转速为1000rpm下搅拌反应2h。tlc监测反应进程,反应结束后得到含有化合物ⅲ的反应液(所述化合物ⅲ的纯度为94.5%);将含有化合物ⅲ的反应液和100ml质量浓度20%的氢氧化钾溶液混合,将ph值调节为中性,在25℃下以1000rpm的搅拌速度搅拌反应1h,tlc监测反应进程,反应结束后,向反应液中加入10g无水硫酸钠进行干燥,旋蒸溶剂得到粗产品,然后将粗产品和20ml乙醇溶液混合进行重结晶,得到邻位环己二酮。所述邻位环己二酮的产率为90.6%,纯度为98.9%;总产率为67.9%。
[0125]
对比例1
[0126]
取4.38g环己酮、10g乙二醇、0.1g对甲苯磺酸和50ml甲苯,在磁力搅拌器下以500rpm的转速搅拌30min使反应液混合均匀,在80℃下回流1h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入1g碳酸钠,搅拌1h,然后倒入100ml水中进行淬灭后,分离得到有机相和水相;将有机相用20ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入50ml乙酸乙酯进行提取,然后向乙酸乙酯中加入3g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为53.6%,纯度为68%。
[0127]
对比例2
[0128]
取4.38g环己酮、10g乙二醇和50ml甲苯,在磁力搅拌器下以500rpm的转速搅拌30min使反应液混合均匀,在80℃下回流1h进行缩合反应;反应完成后,将反应产物冷却至35℃;向反应产物加入1g碳酸钠,搅拌1h,然后倒入100ml水中进行淬灭后,分离得到有机相和水相;将有机相用20ml水进行洗涤,洗涤三次,将洗涤得到的水相和分离得到的水相进行混合后,加入50ml乙酸乙酯进行提取,然后向乙酸乙酯中加入3g无水硫酸钠进行干燥后,旋蒸溶剂得到化合物ⅰ。所述化合物ⅰ的产率为21%,纯度为57%。
[0129]
对比例3
[0130]
在0℃下,将3g化合物ⅰ和20ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加1ml液溴,滴加完毕继续搅拌反应1h。tlc监测反应进程,反应结束向反应液中加入10ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用20ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为53%,纯度为61%。
[0131]
对比例4
[0132]
在80℃下,将3g化合物ⅰ和20ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加1ml液溴,滴加完毕继续搅拌反应1h。tlc监测反应进程,反应结束向反应液中加入10ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用20ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为75%,纯度为86%。
[0133]
对比例5
[0134]
在100℃下,将3g化合物ⅰ和20ml二氯甲烷混合后,在转速为500rpm的搅拌下,以30ml/min的滴加速率向反应液中滴加1ml液溴,滴加完毕继续搅拌反应1h。tlc监测反应进程,反应结束向反应液中加入10ml质量浓度5%的碳酸氢钠溶液进行洗涤,洗涤三次;然后用20ml乙酸乙酯萃取,旋蒸溶剂得到化合物ⅱ。所述化合物ⅱ的产率为73%,纯度为81%。
[0135]
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。