1.本发明涉及降解四环素类物质材料领域,特别是涉及一种应用于光电降解四环素的阳极材料及其制备方法。
背景技术:
2.如今的污水处理工艺中,由于受到经济成本的限制,污水厂大多数都是采用活性污泥法降解有机废水。该方法主要利用污泥的吸附作用和污泥中微生物的代谢作用。四环素等抗生素物质能够对微生物产生直接的毒害,从而造成污泥活性的大幅度降低和维护成本的提高。此外,四环素分子中含有苯环,具有较为稳定的化学结构,所以目前活性污泥法对污水中的四环素的降解效果不甚理想。
3.高级氧化技术主要包括了光化学氧化法、催化湿式氧化法、臭氧氧化法、电化学氧化法、fenton氧化法和类fenton法等。以fenton法氧化降解四环素为例:在实际的处理工艺中,首先将待处理污水的ph值调节到酸性。然后往酸性废水中投加一定浓度的过氧化氢溶液,在光照的条件下,h2o2在存在fe
2+
的条件下,通过链式反应产生氧化性极强的羟基自由基。最后,通过羟基自由基的氧化作用氧化降解四环素。目前,高级氧化技术对四环素的降解具有较好的效率,但其仍旧存在较多的问题。例如,fenton法中的fe
2+
和fe
3+
可以相互转化,但需要外加氧化剂h2o2和调节ph值。其次,fenton法会产生大量含铁污泥,对后续处理工艺以及下一个单元的构筑物带来一定的负担;此外,对于部分难氧化物质,出水浓度并经过处理后还不能达到排放标准。最后,部分高级氧化法还需要高温高压、氧化电压较高等苛刻条件,使得降解成本和环境负担显著增加,以及存在一定的安全隐患等问题,不利于此类高级氧化法在四环素降解领域的大范围推广。
技术实现要素:
4.为解决上述技术问题,本发明提供一种重复利用率高,降解稳定性良好的应用于光电降解四环素的阳极材料及其制备方法。
5.本发明还提供一种应用于光电降解四环素的阳极材料的制备方法,降低了后续处理工艺难度以及减少了材料对下一个单元的构筑物带来的负担。
6.本发明采用如下技术方案:
7.一种应用于光电降解四环素的阳极材料,包括六水氯化铁、硝酸钠、六水氯化钴混合溶液、掺杂氟的sno2导电玻璃。
8.对上述技术方案的进一步改进为,所述阳极材料的制备方法包括如下步骤:将清洗好的fto放置到混合溶液中,通过水热法制得前驱体;然后将前驱体在马弗炉中退火,最终获得co:α-fe2o3阳极材料。
9.一种应用于光电降解四环素的阳极材料的制备方法,包括如下步骤:
10.s1、fto的预处理:截取一定面积的的fto,将fto在有机溶剂和去离子水的分别超声清洗,去除fto上的大部分油污、固体杂质等;随后将fto烘干,得到备用fto;
11.s2、配置反应溶液,其组成为0.10~0.20mol/l的六水氯化铁、1mol/l的硝酸钠和1~50mmol/l六水氯化钴;然后将步骤s1中的备用fto置于聚四氟乙烯的内衬中;根据反应釜的大小,加入反应溶液,密封;
12.s3、将步骤s2中的装好样品的反应釜放入恒温烘箱中,进行水热反应;
13.s4、步骤s3的水热反应结束后取出fto,用有机溶剂和去离子水冲去电极材料表面多余的杂质,烘干;
14.s5、将步骤s4的烘干后的材料导电面朝上,放置在马弗炉中退火,退火操作结束后得到co:α-fe2o3阳极材料。
15.对上述技术方案的进一步改进为,在步骤s1中,所述有机溶剂为丙酮、乙醇、甲醇中的极性溶剂或非极性溶剂。
16.对上述技术方案的进一步改进为,在步骤s1中,所述超声清洗的时间为20分钟。
17.对上述技术方案的进一步改进为,在所述根据反应釜的大小,加入反应溶液,密封步骤中,所述反应溶液的体积用量为反应釜体积的30~80%。
18.对上述技术方案的进一步改进为,在步骤s3中,所述水热反应的温度为90~120℃。
19.对上述技术方案的进一步改进为,在步骤s3中,所述水热反应的反应时间3~12小时。
20.对上述技术方案的进一步改进为,在步骤s5中,所述退火操作的温度为650~850℃,所述退火操作的加热时间为0.5~5小时。
21.对上述技术方案的进一步改进为,在步骤s5中,所述退火操作的升温速度为2~20℃/分钟。
22.本发明的有益效果为:
23.本发明提供一种应用于光电降解四环素的阳极材料,通过co掺杂的方式,制备得到了co:α-fe2o3阳极材料,有效的降低了α-fe2o3的电子-空穴对的复合速率。在光照的条件下,co:α-fe2o3阳极材料可以在较低的电压下实现对盐酸四环素的降解,降低降解成本和提高降解过程的安全性。此外,co:α-fe2o3阳极材料生长在fto上,在完成降解工艺后,可以不用过滤操作,实现对co:α-fe2o3阳极材料的回收,从而降低了后续处理工艺难度以及减少了材料对下一个单元的构筑物带来的负担。
附图说明
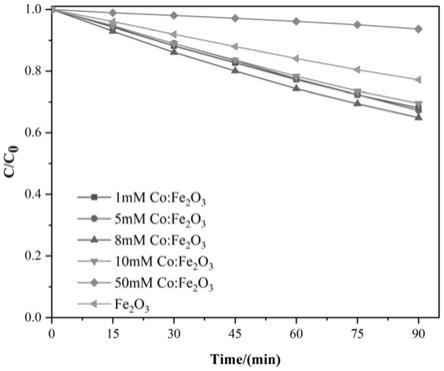
24.图1为本发明的不同掺杂浓度的co:α-fe2o3阳极材料降解四环素效率图;
25.图2为本发明的8mm co:α-fe2o3阳极材料的循环稳定性曲线图;
26.图3为本发明的α-fe2o3阳极材料的扫描电镜图;
27.图4为本发明的8mm co:α-fe2o3阳极材料的扫描电镜图;
28.图5为本发明的8mm co:α-fe2o3阳极材料的x射线衍射图谱。
具体实施方式
29.为更好地理解本发明,下面结合实施例对本发明作进一步说明,但是本发明的实施方式不限于此。
30.本发明中合成用于光电降解盐酸四环素的co:α-fe2o3阳极材料的配方如下所示:0.10~0.20mol/l的六水氯化铁(fecl3·
6h2o)、1mol/l的硝酸钠(nano3)和1~50mmol/l六水氯化钴(cocl2·
6h2o)混合溶液、掺杂氟的sno2导电玻璃(fto)。
31.co:α-fe2o3阳极材料的制备工艺中的各项参数,其中六水氯化钴(cocl2·
6h2o)的添加浓度范围为1~50mmol/l;在该掺杂范围内合成得到的co:α-fe2o3阳极材料对四环素都有更为有益的的降解效率;此外,利用该方法制备得到的co:α-fe2o3阳极材料在实际的降解工艺中,重复利用率高,降解稳定性良好,有着广阔的发展和应用前景。
32.其中,co:α-fe2o3阳极材料是将清洗好的fto放置到混合溶液中,通过水热法制得前驱体;然后将前驱体在马弗炉中退火,最终获得co:α-fe2o3阳极材料。
33.各成分的作用:fto是为电极材料提供生长位点,在降解四环素后进行检测时可以省去过滤的操作,在实际应用中也有降低后续处理成本的作用;在光照和一定的电解质溶液中,co:α-fe2o3阳极材料具有良好的光催化降解盐酸四环素的效果。
34.该co:α-fe2o3阳极材料具体的制备方法为:
35.s1:fto的预处理。截取一定面积的的fto,将fto在有机溶剂和去离子水的分别超声清洗20分钟,去除fto上的大部分油污、固体杂质等,其中有机溶剂可以是丙酮、乙醇、甲醇等极性或非极性溶剂。随后将fto烘干备用。
36.s2:配置反应溶液。其组成为0.10~0.20mol/l的六水氯化铁(fecl3·
6h2o)、1mol/l的硝酸钠(nano3)和1~50mmol/l六水氯化钴(cocl2·
6h2o)。然后将s1中的fto置于聚四氟乙烯的内衬中。根据反应釜的大小,加入反应釜体积30~80%的反应溶液,密封。
37.s3:将装好样品的反应釜放入恒温烘箱中,设定水热反应温度为90~120℃,反应时间3~12小时。
38.s4:水热反应结束后取出fto,用有机溶剂和去离子水冲去电极材料表面多余的杂质,烘干。
39.s5:将s4结束后的材料导电面朝上的放置在马弗炉中,设置退火温度为650~850℃加热0.5~5小时,升温速度为2~20℃每分钟。退火操作结束后得到co:α-fe2o3阳极材料。
40.实施例1:
41.本发明实施例所涉及的co:α-fe2o3合成步骤如下:截取4
×
2.2cm的掺杂氟的sno2导电玻璃(fto),依次用丙酮、乙醇、去离子水超声清洗截取好的fto(丙酮、乙醇、去离子水的超声清洗时间都为20分钟);干燥后备用。
42.称取17g的硝酸钠(nano3),5.406g的六水氯化铁(fecl3·
6h2o),0.380g的六水氯化钴(cocl2·
6h2o)后加入200ml的去离子水搅拌溶解得到混合溶液a(混合溶液a中含有1mol/l的nano3、0.10mol/l的fecl3·
6h2o和0.008mol/l的cocl2·
6h2o)。
43.将干燥后的fto置于25ml的聚四氟乙烯内衬中(注意导电面朝下放置),加入适量的混合溶液a直到恰好淹没整块fto。盖紧聚四氟乙烯内衬盖子后将其放入高温反应釜中,拧紧。最后将反应釜放入烘箱中,设定温度为90℃,加热时间为360min。最终会得到co:α-fe2o3的前驱体。
44.加热完毕后将fto取出,用去离子水冲去fto表面多余的浮沫,然后将冲洗干净的fto放入60℃的烘箱中干燥。完成干燥后用小刀刮去非导电面的薄膜,并且在导电面顶端截取5毫米,也用小刀刮去薄膜,目的是让fto的导电面裸露出一部分,便于后续的电极体系形
成闭合回路。
45.最后将处理好的co:α-fe2o3的前驱体放入马弗炉中,设定升温速率为10℃/min,加热温度为750℃,加热时间为30min。加热完毕后得到掺杂了0.008mol/l的co的α-fe2o3(8mm co:α-fe2o3)。
46.实施例2:
47.本发明中所涉及的1mm co:α-fe2o3、5mm co:α-fe2o3、10mm co:α-fe2o3、50mm co:α-fe2o3、和α-fe2o3都是按照实施例1的步骤所制备的。其中α-fe2o3的合成制备的区别在于没有加入cocl2·
6h2o。
48.实施例3:
49.一个使用co:α-fe2o3阳极材料进行四环素的光电降解过程:选择在356.5nm波长的照射下,记录不同浓度的四环素水溶液的吸光度,根据朗勃-比尔定律定量得到相应的浓度,从而绘制出浓度-吸光度曲线。
50.配制混合溶液b,其中含有40mg/l的盐酸四环素水溶液tch(由于四环素不溶于水,引入盐酸基团后可使得四环素易溶于水)和0.1mol/l的硫酸钠(na2so4)溶液,溶液ph=3.5~4.0。
51.首先构筑三电极体系:阳极为co:α-fe2o3,阴极为铂片,参比电极选用甘汞电极,然后加入100ml混合溶液b后在暗处进行预处理(15min,持续搅拌)以确保四环素水溶液和co:α-fe2o3阳极材料的吸附-脱附平衡,最后在氙灯下模拟太阳光的照射进行光电降解实验。每隔15min取一次样,共取6次,即进行90min的光电降解。取得的样品通过紫外-可见分光光度计测定其四环素的浓度变化,与标准曲线对照可以计算得到降解率为36%。
52.实施例4:
53.co:α-fe2o3阳极材料循环利用光电降解四环素:在第一次循环实验结束后,将co:α-fe2o3阳极材料表面冲洗干净,然后设置与实施例3完全相同的实验条件进行第二次循环实验,以此直至第五次循环实验。第五次循环实验的降解率为33%。
54.如图1所示,相较于纯α-fe2o3,co:α-fe2o3阳极材料对四环素降解效率明显提高。特别的,合成溶液中添加8mmol/l六水氯化钴所制备的8mm co:α-fe2o3阳极材料对四环素的降解效率最高;在较低的外加电压下(0.7v),其降解效率在90min时可以达到35.12%。
55.如图2所示,进行五次平行的循环实验后,优选的8mm co:α-fe2o3阳极材料对四环素的降解效率几乎保持不变,体现了co:α-fe2o3阳极材料具有一定的催化稳定性。
56.从图3和图4的扫描电镜图片可以清晰的观察到,α-fe2o3的纳米结构发生了较为严重的团聚现象,在co:α-fe2o3阳极团聚的较少;其次,优选的8mm co:α-fe2o3阳极材料中co:α-fe2o3纳米颗粒明显小于α-fe2o3纳米颗粒。
57.由图5的x射线衍射图谱可知,co:α-fe2o3阳极主要由sno2和fe2o3构成,其中sno2的衍射峰来自于fto。
58.以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。