1.本发明属于橡胶制品表面处理领域,更具体地说,涉及一种浸胶用的粘合剂、组合物、水分散体、浸胶液、浸胶处理方法及其帘子布。
背景技术:
2.在橡胶制品如轮胎、胶带的生产中,各种纤维骨架材料用来增强橡胶制品的强度。橡胶-织物体系是橡胶制品如轮胎、运输带、三角带等的基础,为了保证聚酰胺、聚酯纤维等织物与橡胶之间具有高的粘合强度,通常,这些纤维材料要经过表面浸胶处理以提高其与橡胶之间的粘合强度。浸胶用粘合剂在作为增强层的轮胎帘子布和具有增强纤维的其它高载荷复合材料中的应用显得尤为重要。众所周知,现有技术中广泛使用间苯二酚甲醛乳胶体系(rfl)来将聚酰胺、聚乙烯以及聚酯纤维等合成纤维粘结到橡胶制品中,尤其是在轮胎帘线领域。
3.聚酰胺(尼龙)纤维只需要通过间苯二酚-甲醛-胶乳(rfl)体系一浴法处理,即可获得理想的粘合效果。然而聚酯纤维材料通常需要经过在间苯二酚-甲醛-胶乳(rfl)体系再附加氯酚树脂或封闭异氰酸酯进行一浴法或二浴法处理。一浴法通常使用氯酚或封闭异氰酸酯与rfl的混合物进行浸胶处理;而要获得更好的粘合效果,通常要使用二浴法浸胶:第一浴使用封闭异氰酸酯和水溶性环氧的组合物进行预处理,第二浴再使用rfl处理。近年来,出于未来环保政策的预测,开始出现rf-free浸胶配方,比较通行的方案是第一浴沿用封闭异氰酸酯和水溶性环氧的组合物预处理,第二浴使用包含封闭异氰酸酯、水性环氧树脂和胶乳的组合物(以下简称rf-free胶液)代替rfl处理。无论是传统的二浴法还是rf-free的二浴法,在聚酯骨架材料的浸胶工艺中,封闭异氰酸酯都是重要的核心原材料之一。
4.橡胶骨架材料浸胶最重要的目的就是通过浸胶使骨架材料与橡胶产生优异的粘合力;为了得到粘结强度更大的封闭异氰酸酯,研究者们进行了大量探索。
5.有研究表明,依赖渗入单元纤维之间的胶粘剂薄膜的机械啮合力,以及弹性体和浸胶液中树脂的官能团与纤维的官能团的分子间作用力和化学作用力,纤维-胶粘剂界面可以达到很高的粘合强度。胶粘剂-橡胶界面的粘合强度在很大程度上还取决于弹性体、胶粘剂和贴胶层共硫化时形成的过渡层的内聚力。
6.一些文献报道了改变浸胶工艺以提升织物与制品之间的粘合力,例如中国专利文献cn108166254a公开了一种高粘合强度聚酯胶管纱的制备方法,该方法通过以马来酸酐代替部分丁吡胶乳作为二次浸胶液进行二次浸胶处理,并且二次浸胶处理采用二段热定型方法,二次浸胶液的成分改变及二段热定型方法对于提高聚酯胶管纱的强度起到了协同作用,使马来酸酐与聚酯工业丝充分接枝,接枝后的聚酯工业丝与epdn树脂的粘合强度提升30%,然而该方法中提高粘合强度的方法并不具有普适性,无法广泛应用于帘子布与橡胶制品的粘合强度的提高。
7.为了提高织物与橡胶的粘合强度,也有较多研究关注于粘合剂配方的改变,例如中国专利文献cn104371091a公开了一种可分散于水中的粉状黏合剂,黏合剂包括部分封端
的低分子异氰酸酯以及作为润湿剂的烷基萘磺酸盐和可能的添加剂,其中为了保证帘线与橡胶的黏着力,控制黏合剂中润湿剂的量不得超过15重量%。然而,润湿剂及其它添加剂的加入在一定程度上仍然会对帘线与橡胶的粘合强度造成一定影响。在帘子布与橡胶制品粘合强度值较低的情况下,例如轮胎或传动带等增强橡胶制品将不能可靠地使用。
8.目前在橡胶骨架材料浸胶中最常用的封闭异氰酸酯基本是以二苯甲烷二异氰酸酯(mdi)为基础,采用己内酰胺、苯酚、丁酮肟等封闭剂进行封闭。目前最常用的是用己内酰胺封闭的mdi,主要产品如瑞士ems公司的il-6系列产品、德国isochen公司的dm-50、国内马鞍山科英公司的cbi-50系列产品;上述牌号的封闭异氰酸酯产品常规粘合强度一般能够满足常规领域中的需求,但对于应用条件更为苛刻的航天领域、高温条件作业环境等,将对橡胶材料的具有更高的性能要求,特别是要求橡胶材料在恶劣、苛刻的条件下具有更加优异的性能,进而使得橡胶材料在恶劣条件下仍具有承压能力、耐冲击、耐疲劳性能。
9.作为浸胶组分,封闭异氰酸酯直接影响织物与橡胶的粘合强度,织物与橡胶粘合强度高的轮胎承压强度高、耐冲击、耐疲劳性能好,适用于如航天等对性能要求较高的领域。因此,如何提高采用封闭异氰酸酯浸胶后的织物与橡胶的粘合强度,是本领域长期致力的焦点问题。
技术实现要素:
10.1.要解决的问题
11.本发明的目的是,寻找一种浸胶用粘合剂,其使橡胶骨架材料与橡胶具有良好的粘合强度,且浸胶后的橡胶骨架材料在恶劣条件下具有更加优异的性能,特别是具有过硫化h抽出力和良好的过硫化保持率。
12.进一步的目的是,提供一种浸胶用粘结剂的制备方法,本发明采用mdi以及papi以一定比例混合作为原料,通过无溶剂的反应获得了在增强嵌入物上、优选在聚酯增强嵌入物、帘子布上具有高过硫化粘结强度保持率的浸胶用粘结剂。
13.进一步的目的是,提供一种上述浸胶用粘合剂为固体成分的水分散体。
14.进一步的目的是,提供一种浸胶方法及其浸胶后的帘子布。
15.2.技术方案
16.为了解决上述问题,本发明所采用的技术方案如下:
17.一种浸胶用的粘合剂,所述粘合剂根据以下规定的高效液相色谱法测定时,色谱图具有己内酰胺封闭的mdi的主峰r1,还包括次色谱峰r2,所述的次色谱峰r2相对于主峰r1的相对保留时间rrt1为1.40~1.43,所述相对保留时间rrt1按照下式计算:
18.rrt1=次色谱峰r2的保留时间/主峰r1的保留时间;
19.高效液相色谱仪:岛津lc-20at;
20.色谱柱:wondasil c18 superb 5μm液相色谱柱;
21.流动相:色谱级四氢呋喃:纯水=50:50;
22.检测波长:260nm;
23.流速:1.1min/ml。
24.本发明虽然限定检测条件为流动相:色谱级四氢呋喃:纯水=50:50,检测波长:260nm,流速:1.1min/ml,但是应当理解的是,设备的系统误差或可以接受的误差应当也是
允许的,而且应当认为与规定的条件实质性相同,同时也应当纳入本专利的保护范围。
25.优选地,所述次色谱峰r2为保留时间为18~22min的色谱峰。
26.优选地,所述次色谱峰r2为保留时间为18~20min的色谱峰。
27.优选地,所述次色谱峰r2相对于主峰r1的相对保留时间rrt1为1.40~1.42。
28.优选地,所述次色谱峰r2与主峰r1的峰面积比rrs1为0~0.5,且次色谱峰r2与主峰r1的峰面积比rrs1不为0;所述峰面积比rrs1按照下式计算:
29.rrs1=次色谱峰r2的峰面积/主峰r1的峰面积。
30.优选地,所述次色谱峰r2与主峰r1的峰面积比rrs1为0.01~0.49。
31.优选地,所述次色谱峰r2与主峰r1的峰面积比rrs1为0.01~0.08。在此峰面积比条件下,显著提高了帘子布或者织物的浸胶后的过硫化保持率,过硫化保持率为将帘子布或者织物处于160℃条件硫化60min的h抽出力与将帘子布或者织物处于160℃条件硫化15min的h抽出力的比值。
32.优选地,所述次色谱峰r2与主峰r1的峰面积比rrs1为0.14~0.49。
33.优选地,所述次色谱峰r2的色谱含量不少于2%,所述次色谱峰r2的色谱含量按下式计算:
34.次色谱峰r2的色谱含量=次色谱峰r2的峰面积/所有峰面积
×
100%。
35.值得说明的是,由于高效液相色谱图中不可避免地会存在杂质峰或溶剂峰,本发明中峰面积占比低于所有峰面积0.1%的色谱峰被认为是不存在,但不影响高效液相色谱图中主峰色谱纯度计算以及次色谱峰色谱含量的计算,即在计算高效液相色谱图中主峰色谱纯度时,仍以包括峰面积占比低于总峰面积0.1%的色谱峰在内的全部可积分的峰面积(所有峰的峰面积,空气峰除外)作为分母;同样地,在计算高效液相色谱图中次色谱峰色谱含量时,仍以包括峰面积占比低于所有峰面积0.1%的色谱峰在内的全部可积分的峰面积(所有峰的峰面积,空气峰除外)作为分母。
36.优选地,所述次色谱峰r2的色谱含量不高于30%。
37.优选地,所述主峰r1色谱纯度为55%~98%;所述主峰r1的色谱纯度按下式计算:
38.主峰r1的色谱纯度=主峰r1的峰面积/所有峰面积
×
100%。
39.优选地,所述主峰r1的色谱纯度为55%~60%。
40.优选地,所述主峰r1的色谱纯度为88%~97%。
41.优选地,所述主峰r1的色谱纯度为92%~97%。
42.优选地,所述主峰r1和次色谱峰r2的色谱纯度或色谱含量计算公式如下:
43.主峰r1色谱纯度=主峰r1的峰面积/所有峰面积
×
100%;
44.次色谱峰r2的色谱含量=次色谱峰r2的峰面积/所有峰面积
×
100%;
45.所述主峰r1与次色谱峰r2的色谱含量之和小于99%。
46.优选地,所述主峰r1与次色谱峰r2的色谱含量之和小于98.5%。
47.优选地,所述主峰r1与次色谱峰r2的色谱含量之和大于88%且小于98.5%。
48.优选地,所述主峰r1与次色谱峰r2的色谱含量之和大于92%且小于98.5%。
49.优选地,所述主峰r1与次色谱峰r2的色谱含量之和为95%~98.5%。
50.优选地,所述主峰r1与次色谱峰r2的色谱含量之和为96%~98.5%。
51.优选地,所述主峰r1与次色谱峰r2的色谱含量之和为70%~87%。
52.优选地,所述主峰r1与次色谱峰r2的色谱含量之和为80%~87%。
53.优选地,所述高效液相色谱进样前,样品配制时,采用25.0mg~30.0mg样品,于105℃下烘干后,溶于40~50ml色谱级四氢呋喃,任意选择地超声30s溶解样品。
54.需要说明的是,此处的烘干是指样品在此温度下干燥至重量不再变化,即烘至恒重或接近于恒重,例如可以是每10min取一次样品,连续三次的称重质量变化在5%以内。
55.值得注意的是,由于仪器测量时不可避免地会产生系统误差,为了尽量减小误差,采用高效液相色谱测量本发明的浸胶用粘合剂时,应当注意以下几个方面:
56.1)在色谱基线平稳后注入样品;
57.2)样品采集结束后色谱工作站自动分析结果;
58.3)样品采集时间为60分钟。
59.一种粘合剂,所述粘合剂根据以下规定的高效液相色谱法测定时,色谱图具有小于或等于480克/摩尔的摩尔质量的封端的低分子量异氰酸酯的主峰r1,还包括次色谱峰r2,所述次色谱峰r2为保留时间为18~20min的色谱峰;
60.高效液相色谱仪:岛津lc-20at;
61.色谱柱:wondasil c18 superb 5μm液相色谱柱;
62.流动相:色谱级四氢呋喃:纯水=50:50;
63.检测波长:260nm;
64.流速:1.1min/ml。
65.优选地,所述小于或等于480克/摩尔的摩尔质量的封端的低分子量异氰酸酯为己内酰胺封闭的mdi。
66.优选地,所述粘合剂根据上述规定的高效液相色谱法测定时,主峰r1为保留时间为13~15min的色谱峰。
67.上述的粘合剂的制备方法,采用mdi与papi混合作为原料,以己内酰胺作为封闭剂,在无溶剂的条件下加热反应至游离异氰酸酯的含量小于1%。
68.优选地,所述papi型号为pm200,所述mdi与papi的质量比为(0.125~20):1。
69.优选地,所述mdi与papi的质量比为(0.125~0.2):1。
70.优选地,所述mdi与papi的质量比为(1~20):1。
71.优选地,所述mdi与papi的质量比为(8~20):1。
72.优选地,所述加热温度为140~160℃。
73.本发明还提供一种水分散体,根据以下规定的高效液相色谱法测定水分散体中的固体组分时,色谱图具有己内酰胺封闭的mdi的主峰r1,还包括次色谱峰r2,所述的次色谱峰r2相对于主峰r1的相对保留时间rrt1为1.40~1.43,所述相对保留时间rrt1按照下式计算:
74.rrt1=次色谱峰r2的保留时间/主峰r1的保留时间;
75.高效液相色谱仪:岛津lc-20at;
76.色谱柱:wondasil c18 superb 5μm液相色谱柱;
77.流动相:色谱级四氢呋喃:纯水=50:50;
78.检测波长:260nm;
79.流速:1.1min/ml;
80.所述高效液相色谱测定前,进样处理为:
81.取所述水分散体40.0mg~65.0mg,于105℃下烘干后,溶于40~50ml色谱级四氢呋喃,任意选择地超声30s溶解样品。此处的烘干定义如前所述。
82.优选地,所述水分散体中的固体组分含量为40%~65%(质量分数)。
83.优选地,所述水分散体中的固体组分含量为40%~55%(质量分数)。
84.需要说明的是,当水分散体中固体组分含量低于40%时,水分散体运输成本较高;当水分散体中固体组分含量高于65%时,由于体系粘度非常高,水分散体制备时加工的流畅性较差、操作困难,使用时也会由于粘度过高而受到限制。综合考虑,水分散体的固体组分含量(质量分数)为40%~65%时属于适宜的含量,尤其是固体组分含量(质量分数)为40%~55%的水分散体。
85.优选地,所述次色谱峰r2为保留时间处于18~22min的色谱峰,优选为保留时间处于18-20min的色谱峰。
86.优选地,所述次色谱峰r2相对于主峰r1的相对保留时间rrt1为1.40~1.42。
87.优选地,所述次色谱峰r2与主峰r1的峰面积比rrs1为0~0.5,且次色谱峰r2与主峰r1的峰面积比rrs1不为0;所述次色谱峰r2与主峰r1的峰面积比rrs1按照下式计算:
88.rrs1=次色谱峰r2的峰面积/主峰r1的峰面积。
89.优选地,所述次色谱峰r2与主峰r1的峰面积比rrs1为0.01~0.49,优选为0.01~0.08,优选为0.14~0.49。
90.优选地,所述水分散体中存在的颗粒的平均粒径小于5μm。此处的平均粒径指的是体积平均粒径d[4,3],采用激光粒度衍射仪以纯水为介质湿法分散检测得到,例如可以采用winner2000zd激光粒度分析仪。
[0091]
优选地,所述水分散体中存在的颗粒的平均粒径小于2.5μm。
[0092]
优选地,所述水分散体中存在的颗粒的d90小于5μm,进一步优选4μm。
[0093]
优选地,所述水分散体中存在的颗粒的d50小于1.2μm。
[0094]
一种组合物,包含前述的粘合剂,所述组合物根据前述规定的高效液相色谱法测定时,具有己内酰胺封闭的mdi的主峰r1,还包括次色谱峰r2,所述的次色谱峰r2相对于主峰r1的相对保留时间rrt1为1.40-1.43,所述相对保留时间rrt1按照下式计算:
[0095]
rrt1=次色谱峰r2的保留时间/主峰r1的保留时间。
[0096]
本发明还提供一种浸胶液,包括前述的水分散体。
[0097]
优选地,还包括环氧树脂、酚醛树脂或胶乳中的一种或几种。
[0098]
优选地,所述浸胶液的固含量为1.5%~25%(质量分数)。
[0099]
一种浸胶处理方法,采用前述所述的粘合剂、水分散体、组合物、浸胶液对橡胶制品用骨架材料进行浸胶处理,所述橡胶制品用骨架材料优选为帘子布。
[0100]
优选地,所述橡胶制品用骨架材料包括聚酯纤维、聚乙烯或聚酰胺中的一种或几种。
[0101]
优选地,采用下述的方式浸胶:
[0102]
(1)一浴胶液:去离子水、环氧树脂以及异氰酸酯粘合剂以一定比例混合作为一浴胶液;
[0103]
(2)二浴胶液:去离子水、片碱、间苯二酚、甲醛、丁吡胶乳、氨水以一定比例混合作
为二浴胶液。
[0104]
需要说明的是,对于本发明使用上述的粘合剂进行二浴法浸胶的方法,在浸胶之前不久,例如可以在浸胶前72h,或浸胶前48h,或浸胶前24h,或浸胶前12h,可通过将一浴胶液和二浴胶液各个组分分别混合而制备一浴胶液和二浴胶液。
[0105]
本发明还提供一种帘子布,采用前述的浸胶处理方法处理得到。
[0106]
3.有益效果
[0107]
相比于现有技术,本发明的有益效果为:
[0108]
(1)本发明的浸胶用的粘合剂、组合物和水分散体,根据本发明规定的高效液相色谱法测定时,具有己内酰胺封闭的mdi的主峰r1,还包括次色谱峰r2,所述的次色谱峰r2相对于主峰r1的相对保留时间rrt1为1.40~1.43,该粘合剂、组合物和水分散体配制为浸胶液后,在增强嵌入物上、优选在聚酯增强嵌入物/橡胶骨架材料上,尤其是增强聚合物产品的轮胎帘子布、输送线绳或v带线绳上,该粘合剂在增强嵌入物和基质(例如橡胶或热塑橡胶或热塑弹性体)之间具有良好的粘合性,具有比己内酰胺封闭的mdi更高的过硫化粘合强度和过硫化保持率;
[0109]
(2)本发明中次色谱峰r2与主峰r1的峰面积比rrs1为0~0.5,不包括0,使得经过本发明的浸胶用的粘合剂、组合物和水分散体浸胶处理后的帘子布具有比己内酰胺封闭的mdi更高的过硫化粘合强度和过硫化保持率;
[0110]
(3)本发明的浸胶用的粘合剂的制备方法,采用mdi与papi混合作为原料,以己内酰胺作为封闭剂,在无溶剂的条件下加热反应至游离异氰酸酯的含量小于1%,相比于传统的mdi结晶法制备的产品,具有更好的过硫化粘合强度和过硫化保持率。
附图说明
[0111]
图1为实施例1中样品ns1的hplc谱图;
[0112]
图2为实施例2-1中样品ns2-1的hplc谱图;
[0113]
图3为对比例b中样品s1的hplc谱图;
[0114]
图4为实施例2-2中样品ns2-2的hplc谱图;
[0115]
图5为实施例2-3中样品ns2-3的hplc谱图;
[0116]
图6为实施例2-4中样品ns2-4的hplc谱图;
[0117]
图7为实施例2-5中样品ns2-5的hplc谱图;
[0118]
图8为实施例2-6中样品ns2-6的hplc谱图;
[0119]
图9为实施例2-7中样品ns2-7的hplc谱图;
[0120]
图10为实施例2-8中样品ns2-8的hplc谱图;
[0121]
图11为实施例2-9中样品ns2-9的hplc谱图;
[0122]
图12为实施例2-10中样品ns2-10的hplc谱图;
[0123]
图13为实施例2-12中样品ns2-12的hplc谱图;
[0124]
图14为对比例c中样品s2的hplc谱图。
[0125]
定义
[0126]
除非另有定义,在本发明中,所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同;本文所使用的术语“和/或”包括一个或多个相关
的所列项目的任意的和所有的组合。
[0127]
在本发明中,实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0128]
在本发明中,织物作为纤维骨架材料用来增强橡胶制品的强度,还可以称之为帘线、帘子布、骨架材料或者嵌入物等。
[0129]
在本发明中,术语“约”用于提供与给定术语、度量或值相关联的灵活性和不精确性。本领域技术人员可以容易地确定具体变量的灵活性程度。
[0130]
在本发明中,“任意选择地”用于表示包括其后描述的方案,或者不包括其后描述的方案,可以根据实际情况选择;例如任意选择地超声30s溶解样品,指代可以选择超声30s溶解样品,当未进行超声时样品已经溶解,也可以不进行超声30s溶解样品的步骤。
[0131]
峰面积比、色谱纯度、色谱含量、温度和其他数值数据可以在本文中以范围格式呈现。应当理解,这样的范围格式仅是为了方便和简洁而使用,并且应当灵活地解释为不仅包括明确叙述为范围极限的数值,而且还包括涵盖在所述范围内的所有单独的数值或子范围,就如同每个数值和子范围都被明确叙述一样。例如,约1至约4.5的数值范围应当被解释为不仅包括明确叙述的1至约4.5的极限值,而且还包括单独的数字(诸如2、3、4)和子范围(诸如1至3、2至4等)。相同的原理适用于仅叙述一个数值的范围,诸如“小于约4.5”,应当将其解释为包括所有上述的值和范围。此外,无论所描述的范围或特征的广度如何,都应当适用这种解释。
[0132]
本发明中,封闭的异氰酸酯指的是官能度大于或等于2的含苯环的异氰酸酯采用封闭剂进行封端的产物。能够作为封闭的异氰酸酯的原料而使用的异氰酸酯结构中包含苯环,另一原料封闭剂的结构中不含有苯环,但其可以为环状结构。作为封闭的异氰酸酯的原料而使用的异氰酸酯,没有特别限定,优选为碳数15~30的异氰酸酯;作为封闭的异氰酸酯的原料而使用的封闭剂,没有特别限定,优选为碳数6~12的酰胺。
[0133]
本发明中,“保留时间”指的是被分离的特定样品组分从进样开始到柱后出现该组分浓度极大值时的时间,也即从进样开始到出现某组分色谱峰的顶点时为止所经历的时间。
[0134]
在本发明中,术语“主峰”,是指反应主产物在高效液相色谱中产生的色谱峰;例如在“对比例”以及在实施例中提及的“主峰”,表示该色谱峰中主成分己内酰胺封闭的mdi所形成的色谱峰,在本发明的色谱测定条件下测试,主峰保留时间约处于13~15min之间。
[0135]
本发明中,峰面积比指的是,本发明实施例获得的样品在进行本发明的hplc测定时,不同保留时间的峰值对应的积分面积的比值,其代表了不同保留时间的物质之间的含量比例;例如,本发明的次色谱峰r2与主峰r1的峰面积比为0~0.5,不包括0,表示存在次色谱峰r2对应的物质,且该物质的含量与主峰r1对应的物质的含量之比为大于0,小于等于0.5。
[0136]
在一些实施方案中,次色谱峰r2与主峰r1的峰面积比可以小于0.5,小于0.48,小于0.43,小于0.42,小于0.27,小于0.20,小于0.15,小于0.10,小于0.09,小于0.08,小于0.05,小于0.04,小于0.03,小于0.02;或者为他们之间的数值,如0.42~0.5,0.27~0.42,0.09~0.15,0.02~0.05。
[0137]
本发明中,相对保留时间指的是,高效液相色谱测定的色谱图中,以特定峰保留时
间为准,计算其他物质色谱峰的相对保留时间,例如以己内酰胺封闭的mdi峰为主峰r1,以主峰r1保留时间为准,次色谱峰r2相对于主峰r1的相对保留时间rrt1,按照下式计算:
[0138]
rrt1=次色谱峰r2的保留时间/主峰r1的保留时间;
[0139]
在本发明中,“色谱纯度”与“色谱含量”计算时,所涉及的“所有峰面积”不计或扣除空气峰,而不认为它是本发明所述各种物质的色谱峰,即不将其计算到“所有峰”之内。
[0140]
相应的,本发明以己内酰胺封闭的mdi的色谱峰为主峰r1;次色谱峰r2指代高效液相色谱中相对于主峰r1的相对保留时间rrt1为1.40~1.43的色谱峰。其前提条件是不计(或者在计算时预先扣除)、空气峰,也不计供试品溶液色谱图中由于测试用试剂(例如溶剂产生的色谱峰,例如配液溶剂与流动相差异)所产生的色谱峰。
[0141]
本发明中,主峰r1的色谱纯度按下式计算:
[0142]
主峰r1的色谱纯度=主峰r1的峰面积/所有峰面积
×
100%。
[0143]
本发明的主峰r1的色谱纯度可以为大于50%,大于55%,大于57%,大于58%,大于60%,大于65%,大于70%,大于74%,大于75%,大于80%,大于83%,大于85%,大于86%,大于89%,大于90%,大于94%,大于95%,大于96%,大于97%,大于98%;或者为他们之间的数值,如50%~55%,60%~65%,70%~75%,80%~85%,90%~95%,96%~98%;或者其它范围,如57.12%~58.22%,74.65%~89.67%,94.68%~96.78%等。
[0144]
本发明中,主峰r1与次色谱峰r2的色谱含量之和指的是己内酰胺封闭的mdi与次色谱峰r2对应的物质的含量之和,例如可以小于99%,小于98%,小于97%,小于96%,小于95%,小于93%,小于90%,小于89%,小于87%,小于85%,小于82%,或者它们之间的范围,97%~99%,85%~98%,85%~99%,82%~85%,或者其它的数值范围,如81.29%~86.35%,88.78%~96.57%,96.80%~98.10%等。
[0145]
本发明中,粘合剂的制备方法,可以采用mdi以及papi作为原料,以己内酰胺作为封闭剂,在无溶剂的条件下加热反应至游离异氰酸酯的含量小于1%,或者小于0.9%,小于0.8%,小于0.7%,小于0.6%,小于0.5%,小于0.4%,小于0.3%,小于0.2%,小于0.1%。原料mdi的比例可以为,0.05%~1%,例如0.05%,0.1%,0.2%,0.3%,0.4%,0.5%,1%;还可以为2%~15%,例如2%,3%,4%,5%,10%,15%;还可以为20%~50%,例如20%,23%,24%,35%,40%,50%;还可以为60%~98%,例如60%,63%,69%,75%,78%,80%,82%,85%,90%,95%,96%,97%,98%;当papi中mdi的含量为20%左右时,原料中加入mdi优选为90%~98%,更优选为95%~98%;当papi中mdi含量为30%左右时,原料中加入mdi优选为85%~95%;当papi中mdi含量为40%左右时,原料中加入mdi优选为80%~93%;当papi中mdi含量达到50%时,原料中可以任意选择mdi,优选为75%~92%,更优选为75%~85%;当papi中mdi含量为60%左右时,原料中可以任意选择加入mdi,其中mdi优选60%~85%,更优选为85%~80%;当papi中mdi含量为70%左右时,原料中可以任意选择mdi,mdi优选40%~60%;当papi中mdi含量为80%左右时,原料中可以任意选择mdi,mdi优选20%~40%;当papi中mdi含量为90%左右时,原料中可以任意选择mdi,mdi优选10%~20%。
[0146]
本发明中,水分散体中存在的颗粒的平均粒径小于10μm,优选为小于5μm。
[0147]
本领域技术人员公知,对于hplc法计算含量而言,可以使用面积归一化计算法,本发明使用的即是面积归一化计算法。另外,即使是采用相同hplc色谱仪、色谱柱以及测定条
件,不同批次的进样也会导致主成分和其它成分的色谱行为会有细微差异,从而导致测定结果有细微差异,这也是本领域技术人员可接受的。
[0148]
本领域技术人员知晓,在色谱条件固定的情况下,物质的色谱行为会相对固定,测定结果之间也会相对固定在一个范围之内;因此对于本发明的封闭的异氰酸酯及组合物、水分散体而言,在本发明规定的色谱条件下,经过多批次测定,主峰r1相对固定在13~15min范围内。
[0149]
当采用其它仪器进行色谱测定时,己内酰胺封闭的mdi的主峰r1和次色谱峰r2的保留时间可能会发生变化,因此,本发明优选在固定的高效液相色谱条件测定。
[0150]
需要特别说明的是,本发明中采用特定高效液相色谱条件下的测定结果来限定粘结剂、组合物及水分散体,是为了更好地确定粘结剂、组合物及水分散体,而特定高效液相色谱条件本身对产品并不构成限制,即并不等于本发明的粘结剂、组合物及水分散体只能采用本发明规定的高效液相色谱来检测;在判断是否为本发明的粘结剂、组合物及水分散体时,如果采用本发明规定的高效液相色谱条件测试的结果落入到本发明保护的范围之内,即可认定其与本发明的粘结剂、组合物及水分散体是相同的。
[0151]
在实验条件不允许时,如果采用其它仪器/条件/参数/色谱柱来检测,那么应当以己内酰胺封闭的mdi作为参考样品进行对照,考察相对保留时间,来判断是否落入本发明保护的范围。
[0152]
在下文的各种试验中,对各种物料(封闭的异氰酸酯、组合物或水分散体等),均采用高效液相色谱测定,具体是按照以下的高效液相色谱法进行,测定方式如下:
[0153]
(1)测试溶液的配制:
[0154]
精密称取(0.0001g天平)待测物料25.0~30.0mg(或水分散体40.0~65.0mg),于105℃下烘干3.5h后,加入40~50ml色谱级四氢呋喃溶解,如有必要,超声30s溶解样品。
[0155]
(2)色谱条件:
[0156]
高效液相色谱仪:岛津lc-20at;
[0157]
色谱柱:wondasil c18 superb 5μm液相色谱柱;
[0158]
流动相:色谱级四氢呋喃:纯水=50:50;
[0159]
检测波长:260nm;
[0160]
流速:1.1min/ml;
[0161]
(3)色谱测试:
[0162]
取待测物料溶液20μl注入液相色谱仪,记录色谱图中主峰、次色谱峰及其它色谱峰的保留时间。
[0163]
(4)色谱计算:
[0164]
以己内酰胺封闭的mdi的峰(例如保留时间处于13~15min)为主峰r1,以主峰r1保留时间为准,次色谱峰r2相对于主峰r1的相对保留时间按照下式计算:
[0165]
rrt1=次色谱峰r2的保留时间/主峰r1的保留时间;
[0166]
次色谱峰r2的保留时间为主峰r1的保留时间数值乘以1.40~1.43。
[0167]
主峰r1色谱纯度按下式计算:
[0168]
主峰r1的色谱纯度=主峰r1的峰面积/所有峰面积
×
100%。
[0169]
次色谱峰r2色谱含量按下式计算:
[0170]
次色谱峰r2的色谱含量=次色谱峰r2的峰面积/所有峰面积
×
100%。
[0171]
实施例中采用的mdi牌号为mdi100,购买自烟台万华聚氨酯股份有限公司;papi牌号为pm200,购买自烟台万华聚氨酯股份有限公司。papi还可以是巴斯夫m20s,亨斯曼5005,拜耳44v20。
[0172]
本发明的粘结强度(或称粘合强度或粘结强力)测试方法如下:
[0173]
一、胶液配制
[0174]
1、一浴胶液的配制
[0175]
采用去离子水、环氧树脂(重量百分比1.0%)、异氰酸酯(重量百分比2.2%)配成一浴胶液。
[0176]
2、二浴胶液的配制
[0177]
采用去离子水、片碱、间苯二酚、甲醛、丁吡胶乳、氨水以如下质量份数配制:2230.0份、2.73份、100.0份、147.3份、2218.0份、120.0份,其中甲醛的固含量为37.0%、丁吡胶乳的固含量为40.4%。
[0178]
二、浸胶实验
[0179]
将帘子布采用一浴胶液浸胶处理,而后采用二浴胶液进行浸胶处理。
[0180]
将经过二浴浸胶处理后的帘子布在160℃下烘干后进行下一步的测试。
[0181]
三、样品性能检测
[0182]
1、帘子布的断裂强力检测
[0183]
2、粘合性能
[0184]
2.1h抽出
[0185]
(1)常规硫化(160℃*15min)粘合强力测试方法为:
[0186]
浸胶帘线按照gb19390-2014所描述的方法(硫化温度与时间不同)测试粘合强力h抽出,常规试验硫化条件为:
[0187]
a)h试片的尺寸为25mm
×
10mm
×
10mm。
[0188]
b)胶料配方(质量份):
[0189]
天然橡胶(1#天然胶)90.0;丁苯橡胶(1500)10.0;硬脂酸(200型,一级)2.0;促进剂dm(优等品)1.2;促进剂tt(优等品)0.03;间接法氧化锌(一级)8.0;n330炭黑35.0;硫磺2.5;粘合剂a0.8;粘合剂rs0.96;总计150.49。
[0190]
c)硫化条件:
[0191]
温度:(160
±
2)℃;
[0192]
时间:15min;
[0193]
对模具施加压力:(3.5
±
0.5)mpa。
[0194]
(2)过硫化(160℃*60min)粘合强力测试方法为:
[0195]
浸胶帘线按照gb19390-2014所描述的方法(硫化温度与时间不同)测试粘合强力h抽出,过硫化试验条件为:
[0196]
a)h试片的尺寸为25mm
×
10mm
×
10mm。
[0197]
b)胶料配方(质量份):
[0198]
天然橡胶(1#天然胶)90.0;丁苯橡胶(1500)10.0;硬脂酸(200型,一级)2.0;促进剂dm(优等品)1.2;促进剂tt(优等品)0.03;间接法氧化锌(一级)8.0;n330炭黑35.0;硫磺
2.5;粘合剂a0.8;粘合剂rs0.96;总计150.49。
[0199]
c)硫化条件:
[0200]
温度:(160
±
2)℃;
[0201]
时间:60min;
[0202]
对模具施加压力:(3.5
±
0.5)mpa。
[0203]
下面结合具体实施例对本发明进一步进行描述,值得说明的是实施例只是具体实施过程中的一个参考示例,并不对本发明的保护范围产生影响。
[0204]
实施例1及其对比例a
[0205]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为10:1
[0206]
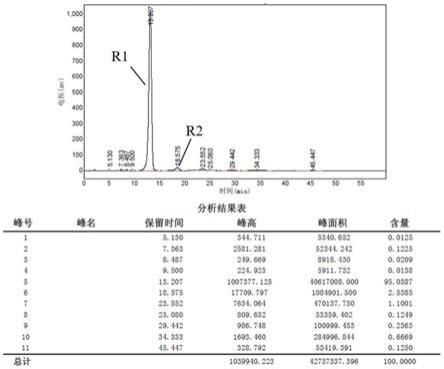
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)160g,papi 16g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns11。将ns11按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图1所示,保留时间在13.207min处的主峰r1色谱纯度为95.04%,保留时间在18.575min(rrt1=1.406)处的次色谱峰r2的含量为2.54%;次色谱峰r2与主峰r1的峰面积比为0.027。
[0207]
以样品ns1为例,本发明中的常规硫化强度、过硫化强度及其它性能参数按照以下试验方法测定:
[0208]
试验线绳:涤纶帘线(厂家a)
[0209]
检测方法:按照同规格帘子布检测方法进行性能检测;
[0210]
试验过程:
[0211]
一、胶液配制
[0212]
1、异氰酸酯粘合剂
[0213]
对比例a:cbi-f为现有技术中的己内酰胺封闭的mdi异氰酸酯粘合剂(来自马鞍山科英合成材料有限公司的市售产品);
[0214]
实施例1:ns1为本实施例1中制备得到的粘合剂ns1的水分散体。
[0215]
2、一浴胶液的配制
[0216]
采用去离子水、环氧树脂(重量百分比1.0%)、异氰酸酯(重量百分比2.2%)配成一浴胶液。其中,异氰酸酯为cbi-f(作为对比例a)或ns1(作为本发明实施例1)。需要说明的是,本实施例中的配比仅作为示例配比,而并非对本发明的限制,对范围的限制仍以权利要求书中的记载为准。在实际配制时,可以采用该配比,也可以不采用该配比。
[0217]
3、二浴胶液的配制
[0218]
采用去离子水、片碱、间苯二酚、甲醛、丁吡胶乳、氨水以如下质量份数配制:2230.0份、2.73份、100.0份、147.3份、2218.0份、120.0份,其中本实施例中甲醛的固含量为37.0%、丁吡胶乳的固含量为40.4%。需要说明的是,本实施例中的配比数据仅作为示例配比,而并非对本发明的限制,对范围的限制仍以权利要求书中的记载为准。在实际配制时,可以采用该配比,也可以不采用该配比。
[0219]
二、浸胶实验
[0220]
将帘子布采用一浴胶液浸胶处理,而后采用二浴胶液进行浸胶处理。
[0221]
将经过二浴浸胶处理后的帘子布在160℃下烘干后进行下一步的测试。
[0222]
三、样品性能检测
[0223]
需要说明的是,由于二浴胶液相同,仅一浴胶液中的异氰酸酯存在差异,以下检测中样品名称为cbi-f指代以cbi-f作为一浴胶液中的异氰酸酯进行浸胶的帘子布(即对比例a),ns1指代以ns1作为一浴胶液中的异氰酸酯进行浸胶的帘子布(即实施例1)。
[0224]
1、帘子布的断裂强力
[0225]
以cbi-f、ns1作为一浴胶液中的异氰酸酯进行浸胶的帘子布的断裂强力分别为215.7n、218.2n。
[0226]
2、粘合性能
[0227]
2.1 h抽出
[0228]
h抽出的测试方法如前所述,测试结果如下表。
[0229]
常规硫化h抽出力(n):
[0230][0231][0232]
值得说明的是,在160℃*15min的常规硫化条件下测量得到的h抽出力(n)都具有较高的h抽出性能,已经满足了工业条件常规的需求;目前行业内着重关注的是特殊、恶劣条件下的如何提高帘子布性能。
[0233]
过硫化h抽出力(n):
[0234]
[0235]
由上述两表格中的数据可知,采用现有cbd-f样品作为一浴胶液的异氰酸酯,h抽出力的力值保持率为71.16%,而采用ns1样品作为一浴胶液的异氰酸酯,h抽出力的力值保持率为82.98%,同时过硫化h抽出力具有优异的性能。,不仅具有优异的强度性能,同时具有优异的强度稳定性。
[0236]
2、耐弯曲疲劳测试
[0237][0238]
由上表中数据可知,相比现有技术中的cbd-f,采用ns1样品作为一浴胶液的异氰酸酯,实验发现在实现断裂强力提高的同时,还保证了强力保持率的提高,具有突破性的进展和令人惊讶的效果。
[0239]
实施例2-1
[0240]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为1.5:1
[0241]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)100g,papi 67g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-1。将ns2-1按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图2所示,保留时间在13.232min处的主峰r1色谱纯度为80.57%,保留时间在18.698min(rrt1=1.413)处的次色谱峰r2的含量为11.72%;次色谱峰r2与主峰r1的峰面积比为0.145;采用涤纶帘线(厂家b)进行常规硫化(160℃,15min)粘结强力为203.7n,过硫化(160℃,60min)粘结强力为181.8n,过硫化保持率为89.25%。
[0242]
对比例b
[0243]
制备封闭的异氰酸酯粘结剂(溶剂法)/100%mdi,溶剂为甲苯
[0244]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入甲苯170g,加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)170g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的晶体记为封闭的异氰酸酯粘结剂s1。将s1按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图3所示,保留时间在13.223min处的主峰r1色谱纯度为98.86%;采用与实施例2-1相同的硫化实验条件测试,常规硫化(160℃,15min)粘结强力为210.3n,过硫化(160℃,60min)粘结强力为173.8n,过硫化保持率为82.64%。
[0245]
实施例2-2
[0246]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为2.5:1
[0247]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)125g,papi 50g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-2。将ns2-2按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图4所示,保留时间在13.165min处的主峰r1色谱纯度为86.94%,保留时间在18.532min(rrt1=1.408)处的次色谱峰r2的含量为8.46%;次色谱峰r2与主峰r1的峰面积比为0.097;采用与实施例2-1相同的硫化实验条件测试,常规硫化(160℃,15min)粘结强力
为202.6n,过硫化(160℃,60min)粘结强力为182.3n,过硫化保持率为89.98%。
[0248]
实施例2-3
[0249]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为3.5:1
[0250]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)140g,papi 40g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-3。将ns2-3按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图5所示,保留时间在13.532min处的主峰r1色谱纯度为89.53%,保留时间在19.198min(rrt1=1.419)处的次色谱峰r2的含量为7.04%;次色谱峰r2与主峰r1的峰面积比为0.079;采用与实施例2-1相同的硫化实验条件测试,常规硫化(160℃,15min)粘结强力为205.4n,过硫化(160℃,60min)粘结强力为185.4n,过硫化保持率为90.26%。
[0251]
实施例2-4
[0252]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为8:1
[0253]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)160g,papi 20g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-4。将ns2-4按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图6所示,保留时间在13.532min处的主峰r1色谱纯度为94.88%,保留时间在19.165min(rrt1=1.416)处的次色谱峰r2的含量为3.22%;次色谱峰r2与主峰r1的峰面积比为0.034;采用与实施例2-1相同的硫化实验条件测试,常规硫化(160℃,15min)粘结强力为207.5n,过硫化(160℃,60min)粘结强力为189.5n,过硫化保持率为91.33%。
[0254]
实施例2-5
[0255]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为10:1
[0256]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)163.5g,papi 16.4g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-5。将ns2-5按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图7所示,保留时间在13.865min处的主峰r1色谱纯度为94.68%,保留时间在19.732min(rrt1=1.423)处的次色谱峰r2的含量为2.12%;次色谱峰r2与主峰r1的峰面积比为0.022;采用与实施例2-1相同的硫化实验条件测试,常规硫化(160℃,15min)粘结强力为199.2n,过硫化(160℃,60min)粘结强力为182.5n,过硫化保持率为91.62%。
[0257]
实施例2-6
[0258]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为1:8
[0259]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)20g,papi 160g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-6。将ns2-6按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图8所示,保留时间在13.157min处的主峰r1色谱纯度为58.22%,保留时间在18.623min(rrt1=1.415)处的次色谱峰r2的含量为28.13%;次色谱峰r2与主峰r1的峰面
积比为0.483;采用与实施例2-1相同的硫化实验条件测试,常规硫化(160℃,15min)粘结强力为209.8n,过硫化(160℃,60min)粘结强力为187.9n,过硫化保持率为89.56%。
[0260]
实施例2-7
[0261]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为1:6
[0262]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)144g,papi 24g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-7。将ns2-7按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图9所示,保留时间在13.328min处的主峰r1色谱纯度为57.12%,保留时间在18.730min(rrt1=1.405)处的次色谱峰r2的含量为24.17%;次色谱峰r2与主峰r1的峰面积比为0.423。采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为89.23%。
[0263]
实施例2-8
[0264]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为1:1
[0265]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)85g,papi 85g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-8。将ns2-8按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图10所示,保留时间在13.047min处的主峰r1色谱纯度为74.65%,保留时间在18.350min(rrt1=1.406)处的次色谱峰r2的含量为14.13%;次色谱峰r2与主峰r1的峰面积比为0.189。采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为89.79%。
[0266]
实施例2-9
[0267]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为2:1
[0268]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)112g,papi 56g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-9。将ns2-9按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图11所示,保留时间在13.075min处的主峰r1色谱纯度为83.50%,保留时间在18.398min(rrt1=1.407)处的次色谱峰r2的含量为8.94%;次色谱峰r2与主峰r1的峰面积比为0.107。采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为89.34%。
[0269]
实施例2-10
[0270]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为4:1
[0271]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)136g,papi 34g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-10。将ns2-10按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图12所示,保留时间在13.158min处的主峰r1色谱纯度为89.67%,保留时间在18.577min(rrt1=1.412)处的次色谱峰r2的含量为5.66%;次色谱峰r2与主峰r1的峰面积比为0.063。采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为90.33%。
[0272]
实施例2-11
[0273]
将实施例1中制备的ns1样品采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为91.25%。
[0274]
实施例2-12
[0275]
制备封闭的异氰酸酯粘结剂(无溶剂法)/mdi:papi约为20:1
[0276]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)160g,papi 8g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂ns2-12。将ns2-12按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图13所示,保留时间在13.198min处的主峰r1色谱纯度为96.78%,保留时间在18.620min(rrt1=1.411)处的次色谱峰r2的含量为1.18%;次色谱峰r2与主峰r1的峰面积比为0.012。采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为90.88%。
[0277]
对比例c
[0278]
制备封闭的异氰酸酯粘结剂(无溶剂法)/纯mdi
[0279]
装有搅拌器、温度计、真空系统的圆底烧瓶中加入二苯基甲烷二异氰酸酯(烟台万华聚氨酯股份有限公司生产的mdi100)167g,加入己内酰胺150g,升温至140℃,搅拌反应,直至取样检测游离异氰酸根含量低于1%,降至常温,得到的黏性固体记为封闭的异氰酸酯粘结剂s2。将s2按照本发明的高效液相色谱条件进行测定,其高效液相色谱如图14所示,保留时间在13.335min处的主峰r1色谱纯度为98.28%。采用与实施例2-1相同的硫化实验条件测试,得到过硫化保持率为82.85%。
[0280]
对比例d
[0281]
采用现有技术中的己内酰胺封闭的mdi异氰酸酯粘合剂cbi-f(来自马鞍山科英合成材料有限公司的市售产品)配制一浴胶液进行与实施例2-1相同条件下的硫化实验,得到常规硫化(160℃,15min)粘结强力为210.5n,过硫化(160℃,60min)粘结强力为174.9n,过硫化保持率为83.09%。
[0282]
由上述的实施例2-1~2-12中,本发明的粘合剂用于配制织物用浸胶液时,相对于现有技术显著地提高了过硫化粘结强力,特别是使得浸胶后织物的过硫化保持率维持在较高的程度,过硫化保持率达到89%以上,显著高于现有技术中粘合剂的过硫化保持率;此外,次色谱峰r2与主峰r1的峰面积比rrs1为0.01~0.08时,过硫化保持率维持在90.26%~91.62%。
[0283]
【水分散体的固含量与粒度的影响】
[0284]
在一些实施例中,将ns2-6配制为固含量(质量含量)为40%的水分散体,该水分散体能够直接用于一浴胶液的配制。在一些实施例中,将ns3配制为固含量(质量含量)为50w%的水分散体,或固含量为55w%的水分散体,或固含量为60w%的水分散体,或固含量为65w%的水分散体,该水分散体能够直接用于一浴胶液的配制。
[0285]
在一些实施例中,将实施例1制备的ns1样品分散于水中,制备成的水分散体中存在的颗粒的平均粒径小于5μm。在一些实施例中,将实施例1制备的ns1样品分散于水中,制备成的水分散体中存在的颗粒的平均粒径小于2.5μm。在一些实施例中,将实施例1制备的ns1样品分散于水中,制备成的水分散体中存在的颗粒的平均粒径小于1.5μm。
[0286]
在一些实施例中,将实施例2-4制备的ns2-4样品分散于水中,制备成的水分散体
中存在的颗粒的d90小于5μm。在一些实施例中,将实施例2-4制备的ns2-4样品分散于水中,制备成的水分散体中存在的颗粒的d90小于4μm。在一些实施例中,将实施例2-4制备的ns2-4样品分散于水中,制备成的水分散体中存在的颗粒的d90小于2.5μm。在一些实施例中,将实施例2-4制备的ns2-4样品分散于水中,制备成的水分散体中存在的颗粒的d50小于1.2μm。
[0287]
将上述粒度的水分散体在30rpm转子、500ml容器条件下测定cbi-f(固含量为50w%)异氰酸酯粘合剂和ns1(50%)(固含量为50w%)异氰酸酯粘合剂的粘度,分别为99.0mpa
·
s和210.0mpa
·
s。针对本发明全新的粘结剂,当控制上述的粒径范围及固含量时,水分散体能够稳定分散、保存和运输,在降低了流转、运输瓶颈与加工流畅性的同时,还能够保证水分散体的稳定性和均一性,使得水分散体配制的浸胶液具有优异的浸胶性能;在该条件下,水分散体可以与织物/橡胶骨架材料充分的接触并渗入织物中,加强浸胶液中的官能团与纤维的官能团的分子间作用力和化学作用力,与现有技术中的水分散体相比其过硫化保持率具有显著地提高。
[0288]
以上内容是对本发明及其实施方式进行了示意性的描述,该描述没有限制性,实施例中所示的也只是本发明的实施方式之一,实际的实施方式并不局限于此。所以,如果本领域的普通技术人员受其启示,在不脱离本发明创造宗旨的情况下,不经创造性的设计出与该技术方案相似的实施方式及实施例,均应属于本发明的保护范围。