一种基于fe4n制备fe4n@pani纳米复合吸波材料的方法
技术领域
1.本发明属于新型功能材料技术领域,涉及一种基于fe4n制备fe4n@pani纳米复合吸波材料的方法。
背景技术:
2.随着电子通讯技术的快速发展和应用,电磁波技术及反电磁波技术被广泛使用。无论是在军事中侦查、隐身技术,或是在日常生活中通讯、电磁波防护。吸波材料在对电磁波吸收、损耗方面表现优异。吸波材料将电磁波反射、吸收转化为其他形式能量进行损耗,如热能。
3.fe4n是一种电子陶瓷材料,具有较高的导电率,稳定的机械性能和化学性能。作为介电损耗型吸波材料,其内部不存在自由载流子,故不会形成感应电流。这类吸波材料的吸波作用为产生极化现象,在外加交变电场下,随着电场周期变化,材料内部的极化行为变化跟不上外场变化,产生极化弛豫,伴随电磁波衰减。但是,单纯的一类吸波材料存在很多不足,往往是阻抗匹配特性与衰减特性之间不能同时兼备,吸收频段窄,材料热稳定性差等。故为了改善材料的吸波性能和其他不足,采用多种吸波机理材料进行复合,不仅可以优化吸波材料的阻抗匹配,还能在不同材料之间产生界面极化效应。
4.聚苯胺(pani)属于导电高分子聚合物,其存在原料易得、制备方法简单、密度小等优点。将pani与无机金属材料复合,可以改善无机金属材料密度过大、结构单一、吸波频段窄等缺点,体现出pani、无机金属共同协作优势:丝滑易加工、增大无机材料分散性、比表面积等。
技术实现要素:
5.为了解决现有技术存在的不足,本发明的目的是提供一种基于fe4n制备fe4n@pani纳米复合吸波材料的方法。基于一种以介电损耗、磁损耗为吸波机理的介电陶瓷吸波材料fe4n为单体,采用原位聚合法将单体与导电高分子聚合物聚苯胺(pani)复合构造界面极化效应的界面,以提高吸波性能和增加有效吸波频宽。
6.本发明提供了一种fe4n@pani纳米复合吸波材料的制备方法,所述制备方法包括如下步骤:
7.步骤一、将三价铁盐溶于有机溶剂中,油浴加热回流获得含铁离子溶胶;
8.步骤二、将步骤一中获得的含铁离子溶胶陈化获得凝胶;
9.步骤三、将步骤二中获得的凝胶高温煅烧获得γ-fe2o3粉末并研磨备用;
10.步骤四、将步骤三所述的γ-fe2o3粉末在真空管式炉中,氮气氛围下升温至500-550℃,转化为在氨气下保温一定时间,再氮气保护降至室温,得到fe4n粉末;
11.步骤五、利用原位聚合法将聚苯胺包覆fe4n纳米颗粒,得到fe4n@pani纳米复合吸波材料。
12.fe4n材料通过溶胶-凝胶法先形成溶胶-凝胶的三维网格结构,在高温失去水分
后,形成前驱体γ-fe2o3粒子,然后采用气体渗氮法获得fe4n材料。
13.步骤一中,所述三价铁盐包括fe(no3)3·
9h2o、fecl3·
6h2o等中的一种或几种;优选地,为fecl3·
6h2o;
14.和/或,所述有机溶剂包括乙二醇、乙二醇/pvp等中的一种或几种;优选地,为乙二醇/pvp;pvp作为表面活性剂、增稠剂,将无机组分与有机溶剂充分混合有利于形成溶胶。
15.和/或,所述三价铁盐和有机溶剂的摩尔比为1:(5-20),具体地,为1:5、1:10、1:15、1:20;为制备出粒径均匀球形γ-fe2o3,优选的摩尔比为1:15。
16.和/或,所述pvp的添加量与fecl3·
6h2o的使用量有关,优选地,添加量为1.0g/0.075mol fecl3·
6h2o。
17.步骤一中,所述油浴温度为70-90℃,所述回流时间为12h;优选地,所述油浴温度为70℃。
18.步骤二中,所述陈化温度为80℃,该温度下能使液相失去流动性,形成凝胶;和/或,所述陈化时间为24h。
19.步骤三中,所述煅烧温度为300-450℃;和/或,所述煅烧升温速率为5℃/min;和/或,所述煅烧时间为2-4h;所述研磨后的粒径要求为20-30nm;优选地,所述煅烧温度为400℃,所述煅烧升温速率为5℃/min,所述煅烧时间为3h;所述粒径要求为24nm。步骤三中的研磨操作能够将煅烧后的大块产物研磨成细小颗粒,使样本更好地取样和保存。陈化操作时烘箱内温度保持稳定,煅烧操作时升温速率适宜是制备获得的γ-fe2o3粒径均匀的重要条件。
20.步骤三中,所述高温煅烧获得的γ-fe2o3为粒径均匀的球形。
21.步骤四中,所述升温速率为3-5℃/min;和/或,所述保温时间为3-5h;优选地,所述升温速率为5℃/min,所述保温时间为4h。
22.步骤四中,所述氮气氛围下优选升温至520℃。
23.步骤五中,所述原位聚合法具体包括如下步骤:称取fe4n、pvp溶于去离子水中,超声,在机械搅拌下滴加一定量苯胺、浓盐酸搅拌;反应后再向溶液中滴加过硫酸铵aps溶液超声反应;最后经离心,水洗、醇洗各三遍得到fe4n@pani复合物。
24.所述fe4n的添加量为0.1g/25ml去离子水;所述pvp的添加量与fe4n的添加量相同;
25.和/或,所述去离子水的用量优选为25ml;
26.和/或,所述溶解后的超声时间为40min-60min;优选地,为40min;
27.和/或,所述苯胺的滴加量为20-200μl;优选地,苯胺的滴加量为100μl;所述浓盐酸为浓度为34%wt-37%wt的分析纯浓盐酸;所述浓盐酸的滴加量优选为50μl;
28.和/或,所述过硫酸铵aps溶液的浓度优选为0.03g/ml;所述过硫酸铵aps溶液的滴加量优选为20ml;所述超声反应的时间优选为2h;
29.和/或,所述醇洗所用的醇为乙醇。
30.由于铁盐与有机溶剂的摩尔比会影响溶胶-凝胶的形成和球形γ-fe2o3的制备,故经过实验,本发明优选摩尔比为1:15的配比进行制备。同时在已有基础上加入pvp修饰铁盐,使溶胶形成得更均匀。对比α-fe2o3。γ-fe2o3有磁性、属于亚稳定态,可以更容易在仅高温分解氨气条件下被[h]还原。在进行复合时,直接在前驱体悬浊液中加盐酸易使盐酸和前驱体反应,故现将fe4n进行修饰后在加入盐酸。调整aps溶液加入时间,试验中以尽量减少
纯pani为目的,加入浓盐酸反应12h后加入过硫酸铵aps溶液,以减少产生纯pani的副产物。尝试将fe4n与pani进行复合探究ani含量对吸波性能的影响。
[0031]
在γ-fe2o3的基础上烧制fe4n,不同的形貌γ-fe2o3得到的fe4n的形貌与对应的γ-fe2o3的形貌基本一致,故球形γ-fe2o3可得到低维化fe4n,有利于吸波性能提高。
[0032]
本发明还提供了上述方法制备获得的fe4n@pani复合物。
[0033]
本发明还提供了上述的fe4n@pani复合物在制备吸波材料中的应用。
[0034]
本发明所述fe4n@pani复合物与现有技术制备的γ-fe2o3@pani的复合物相比,γ-fe2o3作为铁氧体,而fe4n为介电陶瓷材料,两者对吸波的吸收机理不同,一个是通过电阻发热从电变成热,一个是通过磁场弛豫将电磁波消耗掉;本发明所述fe4n@pani复合物的界面极化程度较γ-fe2o3@pani复合物更强,因此介电极化弛豫更强。
[0035]
现有技术中,通过原位聚合法将前躯体γ-fe2o3与pani复合,发现在不同苯胺添加量对复合物的磁性及吸波性能有一定影响。当苯胺添加量为100μl的复合物,该γ-fe2o3@pani复合物在吸波性能方面表现的最佳反射损耗情况为,h=5.5mm时,在频率为7.7ghz时有一个吸波峰值,rl=-12.06db;h=3.0mm时,达到rl(min)=-14.31db,总的有效频宽为6ghz。
[0036]
本发明在制备fe4n@pani复合物时,通过调控苯胺的添加量探究其对fe4n@pani复合物磁性及吸波性能的影响时发现:当苯胺添加量为100μl时,fe4n@pani复合物的反射损耗在频率为9ghz处峰值r
l
为-22.07db,涂层厚度为5.0mm;反射损耗在15.5ghz处峰值为r
l
=-26.15db,涂覆层厚度为3.0mm。
[0037]
在一个具体实施方式中,所述fe4n@pani复合物的制备方法如下:
[0038]
前躯体γ-fe2o3的制备
[0039]
将20.2gfecl3·
6h2o、1.0g pvp溶于40ml的乙二醇,pvp修饰铁盐使其更好地溶于乙二醇形成溶胶,在70℃下油浴加热回流12h,待形成溶胶后,在80℃下陈化24h得到凝胶,将其在马弗炉中高温煅烧得到γ-fe2o3粉末,研碎备用。
[0040]
fe4n的制备
[0041]
将前躯体粉末平铺在瓷舟底部,在真空管式炉中,以5℃/min升温,氮气氛围下升温至520℃,转化为在氨气下保温4h,再以氮气保护降至室温,得到fe4n粉末。
[0042]
fe4n@pani的制备
[0043]
称取0.1gfe4n、0.1g pvp(k-30)溶于25ml去离子水中,超声40min,在机械搅拌下滴加100μl苯胺、50μl浓盐酸搅拌12h。反应后再向溶液中滴加0.03g/ml的过硫酸铵(aps)20ml溶液超声反应2h。最后经离心,水洗、乙醇洗各三遍得到fe4n@pani复合物。
附图说明
[0044]
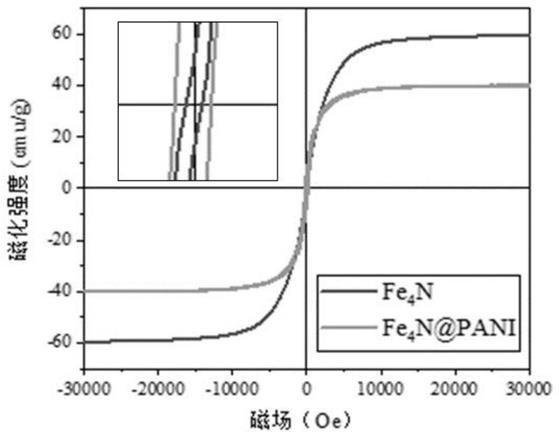
图1为本发明fe4n及fe4n@pani复合物磁滞回线图。
[0045]
图2为本发明苯胺添加量为100μl时fe4n及fe4n@pani复合物反射损耗曲线(左图为fe4n,右图为fe4n@pani复合物)。
[0046]
图3为本发明fe4n及fe4n@pani复合物傅里叶红外光谱曲线。
[0047]
图4是苯胺添加量为50μl时fe4n@pani复合物反射损耗图。
[0048]
图5是苯胺添加量为200μl时fe4n@pani复合物反射损耗图。
[0049]
图6是对比实施例苯胺添加量为100μl时γ-fe2o3@pani复合物反射损耗图。
具体实施方式
[0050]
结合以下具体实施例和附图,对本发明作进一步的详细说明。实施本发明的过程、条件、实验方法等,除以下专门提及的内容之外,均为本领域的普遍知识和公知常识,本发明没有特别限制内容。
[0051]
实施例1
[0052]
前躯体γ-fe2o3的制备
[0053]
将20.2g fecl3·
6h2o、1.0g pvp溶于40ml的乙二醇中,在70℃下油浴加热回流12h,待形成溶胶后,在80℃下陈化24h得到凝胶,将其在马弗炉中高温煅烧得到γ-fe2o3粉末,研碎备用。
[0054]
fe4n的制备
[0055]
将前躯体粉末平铺在瓷舟底部,在真空管式炉中,以5℃/min升温,氮气氛围下升温至520℃,转化为在氨气下保温4h,再以氮气保护降至室温,得到fe4n粉末。
[0056]
fe4n@pani的制备
[0057]
称取0.1gfe4n、0.1g pvp(k-30)溶于25ml去离子水中,超声40min,在机械搅拌下滴加100μl苯胺、50μl浓盐酸搅拌12h。反应后再向溶液中滴加0.03g/ml的过硫酸铵(aps)20ml溶液超声反应2h。最后经离心,水洗、乙醇洗各三遍得到fe4n@pani复合物。
[0058]
性能表征:
[0059]
本发明通过vsm、矢量网络分析进行磁性能表征。
[0060]
a:vsm结果(图1):
[0061]
磁滞回线图可以看出,在振动磁强计中,fe4n有ms=59.3emu/g,通过局部放大得到,mr=1.7emu/g,hc=80.8oe。经过与pani复合后的饱和磁化强度ms降低,而剩余磁化强度mr、矫顽力hc增加,说明添加pani后磁滞现象明显。苯胺添加100μl的fe4n@pani复合物的饱和磁化强度为40.6emu/g、剩余磁化强度mr为8.1emu/g、矫顽力hc为199.8oe。从图中可以看出,fe4n@pani复合物的磁滞回线所围成的面积较fe4n增大,增大其磁滞损耗能力。
[0062]
b:矢量网络分析结果(图2):
[0063]
由图2所示,fe4n最小的反射损耗r
l
在1.0mm、频率为15.34ghz处,其值为-6.42db。fe4n@pani复合物最小反射损耗峰向低频方向移动,在15.5ghz处最小反射损耗值r
l
为-26.15db,涂覆层厚度为3.0mm。中高频段峰值在频率为9ghz处,反射损耗r
l
为-22.07db,涂层厚度为5.0mm。且在该苯胺添加量条件下,频段为6-18ghz均为有效反射损耗值,这大大增加复合物有效频宽。
[0064]
c、傅里叶红外光谱(图3)
[0065]
由图3所示,在已有fe4n的曲线上复合物出现pani的吸收特征峰。在3415cm-1
处为产物内含有一定水分的红外吸收峰外,1580cm-1
为pani中醌式结构振动吸收峰,1495cm-1
为pani中苯式结构振动吸收峰,1302cm-1
为pani中芳香胺吸收峰,1157cm-1
、835cm-1
均为pani中苯环弯曲振动吸收峰。ftir可以确定二者进行复合。
[0066]
实施例2
[0067]
称取0.1gfe4n、0.1g pvp(k-30)溶于25ml去离子水中,超声40min,在机械搅拌下
滴加50μl苯胺、50μl浓盐酸搅拌12h。反应后再向溶液中滴加0.03g/ml的过硫酸铵(aps)20ml溶液超声反应2h。最后经离心,水洗、乙醇洗各三遍得到fe4n@pani复合物。
[0068]
矢量网络分析结果(图4):
[0069]
当苯胺添加量为50μl时,在频率为16ghz处有最小反射损耗,厚度为4.5mm时反射损耗为-15.7db,有效吸波频宽较窄为1ghz。
[0070]
实施例3
[0071]
称取0.1gfe4n、0.1g pvp(k-30)溶于25ml去离子水中,超声40min,在机械搅拌下滴加200μl苯胺、50μl浓盐酸搅拌12h。反应后再向溶液中滴加0.03g/ml的过硫酸铵(aps)20ml溶液超声反应2h。最后经离心,水洗、乙醇洗各三遍得到fe4n@pani复合物。
[0072]
矢量网络分析结果(图5):
[0073]
当苯胺添加量为200μl时,具有两个有效吸波频段分别为8-11ghz、13-18ghz,且在频率为15.6ghz、厚度为3.0mm达到最小反射损耗-16.2db。
[0074]
通过上述不同苯胺添加量数据对比得出,当苯胺添加量为100μl时,复合物的吸波性能表现最好。
[0075]
对比实施例
[0076]
γ-fe2o3@pani的制备
[0077]
称取0.1g γ-fe2o3、0.1g pvp(k-30)溶于25ml去离子水中,超声40min,在机械搅拌下滴加100μl苯胺、50μl浓盐酸搅拌12h。反应后再向溶液中滴加0.03g/ml的过硫酸铵(aps)20ml溶液超声反应2h。最后经离心,水洗、乙醇洗各三遍得到γ-fe2o3@pani复合物。
[0078]
矢量网络分析结果(图6):
[0079]
通过原位聚合法将前躯体γ-fe2o3与pani复合,发现在不同苯胺添加量对复合物的磁性及吸波性能有一定影响。苯胺添加量为100μl的复合物,该复合物在吸波性能方面表现的最佳反射损耗情况为,h=5.5mm时,在频率为7.7ghz时有一个吸波峰值,rl=-12.06db;h=3.0mm时,达到rl(min)=-14.31db,总的有效频宽为6ghz。
[0080]
而本发明在制备fe4n@pani复合物时,通过调控苯胺的添加量探究其对fe4n@pani复合物磁性及吸波性能的影响时发现:当苯胺添加量为100μl时,反射损耗在频率为9ghz处峰值rl为-22.07db,涂层厚度为5.0mm;反射损耗在15.5ghz处峰值为rl=-26.15db,涂覆层厚度为3.0mm。
[0081]
本发明的保护内容不局限于以上实施例。在不背离本发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书为保护范围。