抗细菌化合物
1.本技术是申请日为2016年7月1日,申请号为2016800386447,发明名称为“抗细菌化合物”的发明专利申请的分案申请。
2.本发明涉及新颖的化合物。本发明还涉及此类化合物用于作为药物使用并且进一步用于在治疗细菌性疾病中使用,这些细菌性疾病包括由病原性分枝杆菌(诸如结核分枝杆菌)引起的疾病。此类化合物可以通过干扰结核分枝杆菌中的atp合酶来起作用,其中细胞色素bc1活性的抑制作为主要作用模式。因此,此类化合物主要是抗结核药物。
背景技术:
3.结核分枝杆菌是结核病(tb)的病原体,结核病是一种遍及全世界分布的严重且潜在致命的感染。来自世界卫生组织的估计值指示每年超过800万人感染tb,并且每年200万人死于结核病。在最近十年中,tb病例已经在世界范围增长20%,成为大多数贫穷社区的最重负担。如果这些趋势继续下去,那么tb发病率将在接下来的二十年内增加41%。自从引入一种有效的化学疗法五十年来,tb仍然是位于aids之后的造成世界上成人死亡的主要感染原因。使tb流行病复杂化的是多重耐药株的增多趋势,以及致死性的与hiv的共生。hiv阳性并且感染tb的人比hiv阴性的人多30倍地发展活动性tb的可能性,并且tb是世界范围每三个患有hiv/aids的人中就有一个人死亡的原因。
4.用于治疗结核病的现有方法均涉及多种药剂的组合。例如,由美国公共卫生署推荐的方案是异烟肼、利福平和吡嗪酰胺的组合持续两个月,随后是单独的异烟肼和利福平持续另外的四个月。在感染hiv的患者中,将这些药物继续另外的七个月。对于感染结核分枝杆菌的多重耐药株的患者而言,将药剂,诸如乙胺丁醇、链霉素、卡那霉素、阿米卡星、卷曲霉素、乙硫异烟胺、环丝氨酸、环丙沙星以及氧氟沙星,添加至这些组合疗法中。在结核病的临床治疗中既不存在有效的单一药剂,也不存在提供少于六个月持续时间的疗法的可能性的药剂的任何组合。
5.对于通过实现有助于患者和提供者依从性的方案而改进当前治疗的新药存在高度医学需要。较短的方案以及需要较少监督的那些方案是实现该需要的最佳方式。当一起给予四种药物时,在加强期或杀菌期过程中,来自治疗的大部分益处出现在前2个月;细菌负担大大减少,并且患者变得不再有传染性。需要4个月至6个月的继续或灭菌期来消除持久性杆菌并使复发的风险最小化。将治疗缩短至2个月或更短的有效灭菌药物将是极其有益的。通过需要较少集中监督而有助于依从性的药物也是被需要的。显然,减少治疗的整个长度和药物给予的频率两者的化合物将提供最大益处。
6.使tb流行病复杂化的是多重耐药株或mdr-tb的增加的发病率。世界范围内所有病例的高达百分之四被认为是mdr-tb-耐受四药标准(four-drug standard)中的最有效药物异烟肼和利福平的那些。当不治疗时,mdr-tb是致命的,并且通过标准疗法不能得到充分治疗,所以治疗需要多达2年的“二线”药物。这些药物通常是有毒的、昂贵的且稍微有效的。在缺乏有效疗法的情况下,传染性mdr-tb患者继续传播疾病,用mdr-tb菌株产生新的感染。对于具有新的作用机制的新药存在高度医学需要,该新药可希望对耐药的、特别是mdr菌株展
现活性。
7.如在上文或下文中所使用,术语“耐药”是一个被微生物学的普通技术人员所很好理解的术语。耐药的分枝杆菌是以下分枝杆菌,该分枝杆菌不再易受至少一种先前有效的药物影响;该分枝杆菌已经发展了抵抗被至少一种先前有效的药物的抗生素攻击的能力。耐药菌株可以将该抵抗能力传递给其子代。所述耐受性可以归因于改变其对一种单一药物或对不同药物的敏感性的细菌细胞中的随机遗传突变。
8.mdr结核病是归因于至少耐受异烟肼和利福平的细菌(耐受或不耐受其他药物)的耐药结核病的一种具体形式,异烟肼和利福平是目前两种最强大的抗-tb药物。因此,无论何时在上文或下文中使用,“耐药”包括多重耐药。
9.控制tb流行病的另一个因素是潜伏性tb的问题。不管数十年的结核病(tb)防治规划如何,但是仍有约20亿人无症状地被结核分枝杆菌感染。这些个体中约10%在其寿命期间处于发展为活动性tb的风险中。tb的全球流行被hiv患者由tb的感染以及多药耐受tb菌株(mdr-tb)的出现所激化。潜伏性tb的再活化对于疾病发展而言是一个高风险因素并且导致32%的hiv感染个体死亡。为了控制tb流行病,需要发现可以杀死休眠性或潜伏性杆菌的新药。该休眠的tb可以被再活化以通过若干因素引起疾病,这些因素像如通过使用免疫抑制剂来抑制宿主免疫力,这些免疫抑制剂是像针对肿瘤坏死因子α或干扰素的抗体γ。在hiv阳性患者的情况下,可用于潜伏性tb的唯一预防性治疗是两个月-三个月的利福平、吡嗪酰胺方案。该治疗方案的疗效仍不清楚并且此外,治疗的长度在资源受限的环境中是一种重要约束。因此,对于鉴定可以充当带有潜伏性tb杆菌的个体的化学预防剂的新药存在强烈需要。
10.结核杆菌通过吸入进入健康个体;它们被肺的肺泡巨噬细胞所吞噬。这导致有效的免疫应答以及肉芽肿的形成,肉芽肿由被t细胞包围的结核分枝杆菌感染的巨噬细胞组成。在6-8周的一段时间后,宿主免疫应答通过以下方式导致被感染细胞死亡:被巨噬细胞包围的某些细胞外杆菌、上皮样细胞和周围淋巴组织层坏死和干酪样物质累积。在健康个体的情况下,大部分分枝杆菌在这些环境中被杀死,但小部分杆菌仍存活,并且认为其以非复制、低代谢状态存在且耐受抗-tb药物(例如异烟肼)的杀伤。这些杆菌可以在改变的生理环境中维持甚至持续个体的一生,而不显示疾病的任何临床症状。然而,在10%的这些病例中,这些潜伏性杆菌可以再活化而引起疾病。关于这些顽固性细菌发展的假说之一是人类损害中的病理生理环境,也就是降低的氧张力、营养限制以及酸性ph。已经假定这些因素使得这些细菌对主要的抗分枝杆菌药物显型地有耐药力。
11.除了管理tb流行病之外,还存在耐受一线抗生素的新兴问题。一些重要的实例包括耐青霉素肺炎链球菌、耐万古霉素肠球菌、耐甲氧西林金黄色葡萄球菌、多药耐受沙门氏菌。
12.耐受抗生素的后果是严重的。由耐受性微生物引起的感染不能对治疗做出反应,造成疾病的延长和更大的死亡风险。治疗失败还导致更长期的传染性,这增加了在社区中活动的感染人数,并且因此使一般人群暴露于接触耐受性菌株感染的风险之中。
13.医院是世界范围内抗微生物剂耐受性问题的关键构成。高度易感的患者、集中且延长的抗微生物剂的使用和交互感染的组合已经造成高度耐受性的细菌性病原体的感染。
14.用抗微生物剂自我药疗是引起耐受性的另一主要因素。自我药疗的抗微生物剂可
以是不必要的,常是不适当地给予,或可能不包含适当量的活性药物。
15.患者对推荐治疗的依从性是另一主要问题。患者忘记服药,当他们开始感觉变好时中断了其治疗,或可能不能支付整个疗程,由此创造了微生物适应而非被杀死的理想环境。
16.因为对多重抗生素出现耐受性,医师面临不存在有效疗法的感染。此类感染的发病率、死亡率和财务成本为世界范围的卫生保健系统强加了增加的负担。
17.因此,对于治疗细菌性感染,尤其是分枝杆菌感染(包括耐药性和潜伏性分枝杆菌感染)以及还有其他细菌性感染,尤其是由耐受性细菌菌株引起的那些感染的新的化合物存在高度需要。
18.例如在国际专利申请wo 2011/113606中已经披露了用于治疗结核病的抗感染化合物。这样的文献关注在宿主巨噬细胞内预防结核分枝杆菌增殖的化合物,并且涉及连接(例如通过氨基部分)至例如任选取代的苄基的、具有二环核心的化合物-咪唑并吡啶。
19.国际专利申请wo 2014/015167也披露了被披露为具有治疗结核病的潜在用途的化合物。此处披露的这些化合物具有作为必需元素的二环(5,5-稠合的二环),其被接头基团(例如氨基)取代,所述接头基团本身可附接至另一个二环或芳香族基团。在本文献中的此类化合物不包含一系列的三个以上的环。
20.pethe等人在nature medicine[自然医学],19,1157-1160(2013)中的杂志文章“q203,治疗结核病的有效临床候选药物的发现[discovery of q203,a potent clinical candidate for the treatment of tuberculosis]”中鉴定了针对结核分枝杆菌进行测试的具体化合物。以下描述了化合物q203。
[0021][0022]
这种临床候选药物也在j.medicinal chemistry[医药化学杂志]的杂志文章2014,57(12),第5293-5305页中进行过讨论。据陈述,其具有针对mdr结核病的活性,并且在巨噬细胞内,在mic
50
为0.28nm时,具有针对结核分枝杆菌h37rv的活性。也报道了阳性对照数据(使用已知的抗tb化合物贝达喹啉、异烟肼和莫西沙星)。该文献还建议了基于突变体研究的一种作用方式。它假设其通过干扰结核分枝杆菌中的atp合酶来起作用,并且细胞色素bc1活性的抑制是主要作用方式。细胞色素bc1是atp合成所必需的电子传递链的必要成分。似乎q203对复制和非复制细菌均具有高度活性。
[0023]
国际专利申请wo 2015/014993也披露了具有抗结核分枝杆菌活性的化合物。国际专利申请wo 2013/033070和wo 2013/033167披露了作为激酶调节剂的不同化合物。
[0024]
本发明的目的是提供用于治疗细菌性疾病,尤其是由致病菌如结核分枝杆菌(包括潜伏性疾病和包括耐药性结核分枝杆菌菌株)引起的那些疾病的化合物。此类化合物也是新颖的,并且能够通过干扰结核分枝杆菌中的atp合酶而起作用,其中细胞色素bc1活性
的抑制被认为是主要的作用方式。
技术实现要素:
[0025]
现在提供了具有式(i)的化合物
[0026][0027]
其中
[0028]
r1表示c
1-6
烷基或氢;
[0029]
l1表示接头基团-c(ra)(rb)-(或不存在);
[0030]
x1表示任选的芳香族接头基团;
[0031]
ra和rb独立地表示氢或c
1-6
烷基(任选地被一个或多个氟原子取代);
[0032]
xa表示c(rc)或n;
[0033]
xb表示c(rd)、n、o(在此种情况下,l2不存在)或c=o(在此种情况下,l2也不存在);
[0034]
rc和rd独立地表示h或-ore(其中re表示h或任选地被一个或多个氟原子取代的c
1-6
烷基);
[0035]
q1表示-x
c-(ch2)
n1-x
d-;
[0036]
n1表示0、1或2;
[0037]
q2表示-x
e-(ch2)
n2-x
f-;
[0038]
n2表示0、1或2,但是其中n1和n2不均表示0;
[0039]
xc(其附接至xa)不存在,或者当xa表示ch时,xc可以表示-o-、-nh-或-s-;
[0040]
xd不存在,或者当n1表示2或当xc不存在,xa表示c(rc)并且n1表示1时,xd也可以表示-o-、-nh-或-s-;
[0041]
xe和xf独立地表示不存在,或可以独立地表示-o-、-nh-或-s-,其条件是前述杂原子不直接附接至或α不直接附接至另一个杂原子;
[0042]
q3表示-x
g-(ch2)
n3-x
h-;
[0043]
q4表示-x
i-(ch2)
n4-x
j-;
[0044]
n3表示0、1或2;
[0045]
n4表示0、1或2,但是其中n3和n4不均表示0;
[0046]
xg、xh、xi和xj独立地表示不存在,或可以表示-o-、-nh-或-s-,其条件是前述杂原子不直接附接至或α不直接附接至另一个杂原子;
[0047]
当xb表示o或c=o时,l2不存在;
[0048]
当xb表示c(rd)(例如ch)或n时,l2可以表示氢、卤素、-orf、c
1-6
烷基(任选地被一个或多个卤素(例如氟原子)取代)或芳香族基团(任选地被一个或多个选自卤素、c
1-6
烷基(其
本身任选地被一个或多个选自氟、-cf3和/或-sf5的取代基取代)、-oc
1-6
烷基(其本身任选地被一个或多个氟原子取代)、-o-苯基(其本身任选地被卤素、c
1-6
烷基、c
1-6
氟烷基和/或-oc
1-6
烷基取代)或-sf5的取代基取代);
[0049]
rf表示氢或c
1-6
烷基(任选地被一个或多个氟取代);
[0050]
环a是包含至少一个杂原子(优选包含至少一个氮原子)的5元芳香族环;
[0051]
环b是5或6元环,其可以是芳香族的或非芳香族的,任选地包含1至4个杂原子(优选地选自氮、氧和硫);
[0052]
环a和/或环b可以任选地被一个或多个选自以下的取代基取代:卤素、c
1-6
烷基(任选地被一个或多个卤素(例如氟原子)取代)和/或-oc
1-6
烷基(其本身任选地被一个或多个氟原子取代),
[0053]
或其药学上可接受的盐;
[0054]
该化合物在此处可以被称为“本发明的化合物”。
[0055]
具体而言,在本发明的主要实施例中,提供了以下具有式(ia)的化合物,用于在结核病的治疗中使用:
[0056][0057]
其中
[0058]
r1表示c
1-6
烷基或氢;
[0059]
l1表示接头基团-c(ra)(rb)-;
[0060]
x1表示任选的碳环芳香族接头基团(该接头基团本身可以任选地被一个或多个取代基取代,该一个或多个取代基选自氟、-oh、-oc
1-6
烷基和c
1-6
烷基,其中后两个烷基部分本身任选地被一个或多个氟原子取代);
[0061]
ra和rb独立地表示氢或c
1-6
烷基(任选地被一个或多个氟原子取代);
[0062]
xa表示c(rc)或n;
[0063]
xb表示c(rd)、n、o(在此种情况下,l2不存在)或c=o(在此种情况下,l2也不存在);
[0064]
rc和rd独立地表示h、f或-ore(其中re表示h或任选地被一个或多个氟原子取代的c
1-6
烷基,或者,rd和l2可以连接到一起以形成任选地包含1至3个杂原子的4至6元的环状基团(即螺环);
[0065]
q1表示-x
c-(ch2)
n1-x
d-;
[0066]
n1表示0、1或2;
[0067]
q2表示-x
e-(ch2)
n2-x
f-;
[0068]
n2表示0、1或2,但是其中n1和n2不均表示0;
[0069]
xc(其附接至xa)不存在,或者当xa表示ch时,xc可以表示-o-、-nh-或-s-;
[0070]
xd不存在,或者当n1表示2或当xc不存在,xa表示c(rc)并且n1表示1时,xd也可以表
示-o-、-nh-或-s-;
[0071]
xe和xf独立地表示不存在,或可以独立地表示-o-、-nh-或-s-,其条件是前述杂原子不直接附接至或α不直接附接至另一个杂原子;
[0072]
q3表示-x
g-(ch2)
n3-xhh-;
[0073]
q4表示-x
i-(ch2)
n4-x
j-;
[0074]
n3表示0、1或2;
[0075]
n4表示0、1或2,但是其中n3和n4不均表示0;
[0076]
xg、xh、xi和xj独立地表示不存在,或可以表示-o-、-nh-或-s-,其条件是前述杂原子不直接附接至或α不直接附接至另一个杂原子;
[0077]
当xb表示o或c=o时,l2不存在;
[0078]
当xb表示c(rd)(例如ch)或n时,l2可以表示氢、卤素、-orf、-c(o)-rg、c
1-6
烷基(任选地被一个或多个卤素(例如氟原子)取代)或芳香族基团(任选地被一个或多个取代基取代,该一个或多个取代基选自卤素、c
1-6
烷基(其本身任选地被一个或多个取代基取代,该一个或多个取代基选自氟、-cf3和/或-sf5)、-oc
1-6
烷基(其本身任选地被一个或多个氟原子取代)、-o-苯基(其本身任选地被卤素、c
1-6
烷基、c
1-6
氟烷基和/或-oc
1-6
烷基取代)或-sf5);
[0079]
rf表示氢、c
1-6
烷基(任选地被一个或多个氟取代)或芳香族基团(其本身任选地被一个或多个取代基取代,该一个或多个取代基选自卤素、c
1-6
烷基和-oc
1-6
烷基,其中后两个烷基部分本身可以任选地一个或多个氟原子取代);
[0080]
rg表示氢或c
1-6
烷基(任选地被一个或多个取代基取代,该一个或多个取代基选自氟、或-oc
1-3
烷基,其中后一个部分也任选地被一个或多个氟原子取代)或芳香族基团(任选地被一个或多个取代基取代,该一个或多个取代基选自卤素、c
1-6
烷基或-oc
1-6
烷基);
[0081]
环a可以通过虚线表示的两个可能的键中的一个附接至必需的酰胺部分(即-c(o)-n(r1)-部分),这两个可能的键在(该环的)两个不同的原子处连接至环a;
[0082]
环a是包含至少一个杂原子(优选包含至少一个氮原子)的5元芳香族环;
[0083]
环b是5或6元环,其可以是芳香族的或非芳香族的,任选地包含1至4个杂原子(优选地选自氮、氧和硫);
[0084]
环a和/或环b可以任选地被一个或多个选自以下的取代基取代:卤素、c
1.6
烷基(任选地被一个或多个卤素(例如氟原子)取代)和/或-oc
1-6
烷基(其本身任选地被一个或多个氟原子取代),
[0085]
或其药学上可接受的盐,
[0086]
该化合物在此处也可以被称为“本发明的化合物”。
[0087]
例如,具有式(ia)的化合物可以如上所述,可以使得环a经由特定的环原子连接至酰胺部分,如以下具有式(i)的化合物所描绘:
[0088][0089]
该实施例实质上是环a经由式(ia)中虚线之一所表示的键连接至必需的氨基部分上的图示。
[0090]
药学上可接受的盐包括酸加成盐和碱加成盐。可以通过常规手段,例如,通过将具有式i的化合物的游离酸或游离碱形式与一个或多个当量的适当的酸或碱、任选地在溶剂中或在该盐不可溶于其中的介质中进行反应,随后使用标准技术(例如,在真空中,通过冷冻干燥或通过过滤)去除所述溶剂或所述介质来形成此类盐。还可以通过将处于盐形式的本发明的化合物的一种反离子与另一种反离子进行交换,例如使用一种适合的离子交换树脂,来制备多种盐。
[0091]
如上文所提及的药学上可接受的酸加成盐意指包括具有式(i)的化合物所能形成的治疗活性的无毒酸加成盐形式。这些药学上可接受的酸加成盐可以方便地通过用这种适当的酸来处理碱形式来获得。适当的酸包括例如,无机酸,如氢卤酸(例如盐酸或氢溴酸)、硫酸、硝酸、磷酸以及类似酸;或有机酸,例如像乙酸、丙酸、羟基乙酸、乳酸、丙酮酸、草酸(即乙二酸)、丙二酸、琥珀酸(即丁二酸)、马来酸、富马酸、苹果酸、酒石酸、柠檬酸、甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸、环己氨基磺酸、水杨酸、对氨基水杨酸、双羟萘酸以及类似酸。
[0092]
出于本发明的目的,本发明化合物的溶剂化物、前药、n-氧化物和立体异构体也包括在本发明的范围内。
[0093]
本发明的相关化合物的术语“前药”包括任何化合物,其在口服或肠胃外给药后,在体内被代谢以形成实验上-可检测的量的化合物,并且是在预定的时间(例如在6和24小时之间的给药间隔(即每天一次至四次))之内。为免生疑问,术语“肠胃外的”给药包括除了口服给药外所有的给药形式。
[0094]
本发明的化合物的前药可以按以下的方式通过修饰存在于该化合物上的官能团来制备,该方式使得当向哺乳动物受试者给予此类前药时,这些修饰在体内被切割。典型地,通过合成具有前药取代基的母体化合物来完成这些修饰。前药包括本发明的化合物,其中在本发明的化合物中的羟基、氨基、巯基、羧基或羰基基团被结合到在体内可以被切割的任何基团上以分别再生出游离的羟基、氨基、巯基、羧基或羰基基团。
[0095]
前药的实例包括但不局限于,羟基官能团的酯和氨基甲酸酯、羧基官能团的酯基、n-酰基衍生物和n-曼尼希碱。有关前药的一般信息可以例如在班德加德(bundegaard,h.)“前药的设计”第1-92页,爱思唯尔(elesevier),纽约-牛津(new york-oxford)(1985)中找到。
[0096]
本发明的化合物可以包含双键并且因此可以存在为关于每个单独双键的e(异侧)和z(同侧)几何异构体。位置异构体也可以被包括在本发明的这些化合物中。所有此类异构
体(例如,如果本发明的化合物包含一个双键或稠环,则包括顺式和反式形式)及其混合物都包括在本发明的范围之内(例如,单一的位置异构体和位置异构体的混合物都可以包括在本发明的范围之内)。
[0097]
本发明的化合物还可以展示出互变异构现象。所有的互变异构形式(或互变异构体)及其混合物都包括在本发明的范围之内。术语“互变异构体”或“互变异构形式”指的是具有不同能量的结构异构体,这些异构体可经由低能量势垒相互转换。例如,质子互变异构体(也称作质子移变互变异构体)包括经由质子移变产生的相互转换,诸如酮-烯醇和亚胺-烯胺异构化。价键互变异构体包括由一些成键电子的重组产生的相互转换。
[0098]
本发明的化合物还可以包含一个或多个不对称碳原子并且因此可以展示出旋光和/或非对映异构现象。可以使用常规技术,例如,层析法或分步结晶来分离非对映异构体。可以通过使用常规技术,例如分步结晶或hplc,对这些化合物的外消旋混合物或其他混合物进行分离来分选不同的立体异构体。可替代地,所希望的旋光异构体可以通过适当的旋光起始材料在不会引起外消旋作用或差向异构作用(epimerisation)的条件(即一种
‘
手性池’(
‘
chiral pool’)方法)下的反应;通过衍生化作用(即,拆分,包括动态拆分)适当的起始材料与一种可以在适合的阶段被去除的
‘
手性助剂’(例如与一种纯手性酸)反应,随后通过常规手段(例如层析法)分离非对映异构体衍生物;或者通过与一种适当的手性试剂或手性催化剂反应来制备,所有都在本领域普通技术人员已知的条件下。
[0099]
所有的立体异构体(包括但不局限于非对映异构体、对映异构体和阻转异构体)及其混合物(例如,外消旋混合物)都包含在本发明的范围之内。
[0100]
在此示出的这些结构中,在任何具体的手性原子的立体化学都未指明的情况下,那么所有的立体异构体都被认为是本发明的化合物并且包括在本发明的化合物中。在立体化学通过表示一个具体构型的实楔形线或虚线被指明的情况下,那么该立体异构体是所指明和定义的。
[0101]
本发明的这些化合物能以非溶剂化的形式连同与药学上可接受的溶剂(诸如水、乙醇等)的溶剂化的形式存在,并且意在表明本发明包括溶剂化的以及非溶剂化的形式两者。
[0102]
本发明还包括本发明的同位素标记的化合物,这些同位素标记的化合物与在此列举的那些相同,但是事实上一个或多个原子由具有原子质量或质量数不同于自然中通常发现(或自然中发现的最多的那一个)的原子质量或质量数的原子所代替。在此指明的任何具体的原子或元素的所有同位素都被认为是在本发明的这些化合物的范围之内。可以包含在本发明化合物中的示例性同位素包括氢、碳、氮、氧、磷、氟、氯和碘的同位素,诸如2h、3h、
11
c、
13
c、
14
c、
13
n、
15
o、
17
o、
18
o、
32
p、
33
p、
35
s、
18
f、
36
cl、
123
i、及
125
i。本发明的某些同位素标记的化合物(例如,用3h和
14
c标记的那些)在化合物中是有用的并且用于底物组织分布测定。氚化(3h)和碳-l4(
14
c)同位素是有用的,因为它们使得制备和可检测性变得容易。此外,用更重同位素(诸如氘)(即,2h)取代可以提供由于更大的代谢稳定性而产生的某些治疗优点(例如,增加的体内半衰期或降低的剂量需求)并且因此在一些环境下可以是优选的。正电子发射同位素(诸如
15
o、
13
n、
11
c和
18
f)对于正电子发射断层术(pet)研究是有用的以检查底物受体的占用率。一般可以通过与在说明书/或在下文的实例中描述的那些类似的以下程序、通过用同位素标记的试剂取代非同位素标记的试剂来制备本发明的同位素标记的化合物。
[0103]
除非另外指明,在此定义的c
1-q
烷基基团(其中q是该范围的上限)可以是直链的,或者,当存在足够数目(即,如果适当的话,最少两个或三个)的碳原子时,可以是支链的和/或环的(这样即形成c
3-q-环烷基基团)。此类环烷基基团可以是单环的或二环的并且可以进一步是桥接的。此外,当存在足够数目(即,最少四个)的碳原子时,此类基团还可以是部分环的。此类烷基基团还可以是饱和的,或者当存在足够数目(即,最少两个)的碳原子时,可以是不饱和的(例如,形成c
2-q
烯基或c
2-q
炔基基团)。
[0104]
可以被特别提及的c
3-q
环烷基基团(其中q是该范围的上限)可以是单环的或二环的烷基基团,该环烷基基团可以进一步是桥接的(这样即形成,例如,稠环系统,诸如三个稠合的环烷基基团)。此类环烷基基团可以是饱和的或包含一个或多个双键的不饱和的(例如,形成环烯基基团)。多个取代基可以附接在该环烷基基团上的任何位点处。此外,在存在足够数目(即,最少四个)的碳原子的情况下,此类环烷基基团还可以是部分环的。
[0105]
术语“卤素”,当在此使用时,优选包含氟、氯、溴和碘。
[0106]
杂环基团(当在此提及时)可以包括芳香族或非芳香族杂环基团,并且因此包括杂环烷基和杂芳基。同样地,“芳香族或非芳香族5或6元环”可以是在环中具有5或6个成员的杂环基团(以及碳环基团)。
[0107]
可以被提及的杂环烷基基团包括非芳香族单环和二环的杂环烷基基团,其中在该环系统中的这些原子中的至少一个(例如,一至四个)是除碳外(即一个杂原子),并且其中在该环系统中的原子的总数在3与20之间(例如,在三和十之间,例如,在3和8之间,诸如5-至8-)。此类杂环烷基基团还可以是桥接的。此外,此类杂环烷基基团可以是饱和的,或包含一个或多个双键和/或三键的不饱和的,从而形成,例如,c
2-q
杂环烯基(其中q是该范围的上限)。可以被提及的c
2-q
杂环烷基基团包括7-氮杂二环[2.2.1]庚基、6-氮杂二环[3.1.1]庚基、6-氮杂二环[3.2.1]-辛基、8-氮杂二环-[3.2.1]辛基、吖丙啶基、氮杂环丁烷基、二氢吡喃基、二氢吡啶基、二氢吡咯基(包括2,5-二氢吡咯基)、二氧戊环基(包括1,3-二氧戊环基)、二噁烷基(包括1,3-二噁烷基和1,4-二噁烷基)、二噻烷基(包括1,4-二噻烷基)、二硫戊环基(包括1,3-二硫戊环基)、咪唑烷基、咪唑啉基、吗啉基、7-氧杂二环[2.2.1]庚基、6-氧杂二环-[3.2.1]辛基、氧杂环丁烷基、环氧乙烷基、哌嗪基、哌啶基、非芳香族吡喃基、吡唑烷基、吡咯烷酮基、吡咯烷基、吡咯啉基、奎宁环基、环丁砜基、3丁二烯砜基、四氢吡喃基、四氢呋喃基、四氢吡啶基(诸如1,2,3,4-四氢吡啶基和1,2,3,6-四氢吡啶基)、硫杂环丁烷基、硫杂环丙烷基、硫杂环戊烷基、硫代吗啉基、三噻烷基(包括1,3,5-三噻烷基)、托烷基等。在适当的情况下,杂环烷基基团上的取代基可以位于该环系统中的任何原子(包括杂原子)上。杂环烷基基团的附接点可以是经由该环系统中的任何原子(在适当的情况下),包括杂原子(诸如氮原子),或在可以作为该环系统的部分存在的任何稠合的碳环上的原子。杂环烷基还可以处于n-或s-氧化的形式。在此提及的杂环烷基可以被确切地指定为单环的或二环的。
[0108]
芳香族基团可以是芳基的或杂芳基的。可以被提及的芳基基团包括c
6-20
,诸如c
6-12
(例如,c
6-10
)芳基基团。此类基团可以是单环的、二环的或三环的并且具有6与12(例如,6与10)个之间的环碳原子,其中至少一个环是芳香族的。c
6-10
芳基包括苯基、萘基以及类似基团,诸如1,2,3,4-四氢萘基。芳基基团的附接点可以是经由该环系统的任何原子。例如,当该芳基基团是多环的时候,该附接点可以是经由原子,包括非芳香族环的原子。然而,当芳
基基团是多环(例如,二环或三环)的时候,它们优选地是经由一个芳香族环连接到该分子的其余部分上。在此可以被提及的最优选的芳基基团是“苯基”。
[0109]
除非另外说明,术语“杂芳基”当在此使用时指的是包含一个或多个杂原子(例如一个至四个杂原子)的芳香族基团,该一个或多个杂原子优选地选自n、o和s。杂芳基包括具有5和20元之间(例如,5和10元之间)的那些,并且可以是单环的、二环的或三环的,其条件是这些环中至少一个是芳香族的(这样形成,例如,一个单-、二-或三环的杂芳基)。当该杂芳基基团是多环的时,该附接点可以是经由任何原子,包括非芳香族环的原子。然而,当杂芳基基团是多环(例如,二环或三环)的时,它们优选地是经由一个芳香族环连接到该分子的其余部分上。可以被提及的杂芳基包括3,4-二氢-1h-异喹啉基、1,3-二氢异吲哚基、1,3-二氢异吲哚基(例如,3,4-二氢-1h-异喹啉-2-基、1,3-二氢异吲哚-2-基、1,3-二氢异吲哚-2-基;即,经由一个非芳香族环连接的杂芳基),或者,优选地是,吖啶基、苯并咪唑基、苯并二噁烷基、苯并二氧杂环庚基、苯并二氧杂环戊烯基(包括1,3-苯并二氧杂环戊烯基)、苯并呋喃基、苯并呋吖基、苯并噻二唑基(包括2,1,3-苯并噻二唑基)、苯并噻唑基、苯并噁二唑基(包括2,1,3-苯并噁二唑基)、苯并噁嗪基(包括3,4-二氢-2h-1,4-苯并噁嗪基)、苯并噁唑基、苯并吗啉基、苯并硒杂二唑基(包括2,1,3苯并硒杂二唑基)、苯并噻吩基、咔唑基、色满基、噌啉基、呋喃基、咪唑基、咪唑并[1,2-a]吡啶基、吲唑基、二氢吲哚基、吲哚基、异苯并呋喃基、异色满基、异二氢吲哚基、异吲哚基、异喹啉基、异噻唑基、异硫代苯并二氢吡喃基、异噁唑基、萘啶基(包括1,6-萘啶基或者,优选地是,1,5萘啶基和1,8-萘啶基)、噁二唑基(包括1,2,3-噁二唑基、1,2,4噁二唑基和1,3,4噁二唑基)、噁唑基、吩嗪基、吩噻嗪基、酞嗪基、蝶啶基、嘌呤基、吡喃基、吡嗪基、吡唑基、哒嗪基、吡啶基、嘧啶基、吡咯基、喹唑啉基、喹啉基、喹嗪基、喹喔啉基、四氢异喹啉基(包括1,2,3,4-四氢异喹啉基和5,6,7,8-四氢异喹啉基)、四氢喹啉基(包括1,2,3,4-四氢喹啉基和5,6,7,8-四氢喹啉基)、四唑基、噻二唑基(包括1,2,3-噻二唑基、1,2,4-噻二唑基和1,3,4-噻二唑基)、噻唑基、硫代苯并二氢吡喃基、硫代乙氧苯基、噻吩基、三唑基(包括1,2,3-三唑基、1,2,4-三唑基和1,3,4-三唑基)以及类似基团。在适当的情况下,杂芳基基团上的取代基位于该环系统中的任何原子(包括杂原子)上。杂芳基基团的附接点可以是经由该环系统中的任何原子(在适当的情况下),包括杂原子(诸如氮原子),或在可以作为该环系统的部分存在的任何稠合的碳环上的原子。杂芳基还可以处于n-或s-氧化的形式。在此提及的杂芳基基团可以被确切地指定为单环的或二环的。当杂芳基基团是其中存在一个非芳香族环的多环时,该非芳香族环可以由一个或多个=o基团取代。在此可以被提及的最优选的杂芳基基团是包含1、2或3个杂原子(例如优先选自氮、氧和硫)的5-或6元芳香族基团。
[0110]
可以特别指出地是,该杂芳基基团是单环的或二环的。在指定该杂芳基为二环的情况下,那么它可以由与另一个五-、六-或七-元的环(例如,一个单环的芳基或杂芳基环)稠合的一个五-、六-或七-元的单环(例如,一个单环的杂芳基环)组成。
[0111]
可以被提及的杂原子包括磷、硅、硼,并且优选地是氧、氮和硫。
[0112]
当在此处中提到“芳香族”基团时,它们可以是芳基的或杂芳基的。当在此处提及“芳香族接头基团”时,它们可以是如此处所定义的芳基或杂芳基,优选是单环的(但可以是多环的)并且经由该接头基团的任何可能的原子连接到分子的其余部分。然而,当具体涉及碳环芳香族接头基团时,这样的芳香族基团可以不含杂原子,即它们可以是芳基(但不是杂
芳基)。
[0113]
为免生疑问,在此指出一个基团可以由一个或多个取代基(例如,选自c
1-6
烷基)取代的情况下,那么这些取代基(例如烷基基团)是彼此独立的。即,此类基团可以由相同的取代基(例如相同的烷基取代基)或不同的(例如烷基)取代基取代。
[0114]
在此提及的所有个体特性(例如,优选特征)可以独立地或与在此提及的任何其他特征(包括优选特征)组合地采用(因此,优选特征可以与其他优选特征结合或独立于它们地采用)。
[0115]
技术人员将理解为本发明主题的本发明的化合物包括稳定的那些。即,本发明的化合物包括足够稳健以承受从例如一种反应混合物分离至有用纯度的那些。
[0116]
如上所述,在本发明的主要实施例中,本发明的化合物是如下化合物,在这些化合物中:
[0117]
l1表示接头基团-c(ra)(rb)-;并且
[0118]
x1表示任选的碳环芳香族接头基团;并且
[0119]
具有式(ia)的化合物代表具有式(i)的化合物。
[0120]
下文中描述的优选的化合物或其他方面或实施例可以涉及本发明的这种主要实施例(在这种情况下,l1或x1的不一致的定义是多余的),其中l1和/或x1的这种定义可以与一个或多个其他特征或方面(例如在下文中描述的那些,诸如所描述的一些优选方面)组合地采用。
[0121]
本发明的优选化合物包括那些,其中:
[0122]
当xa表示c(rc)时,它优选地是ch;
[0123]
xa表示ch或n;
[0124]
re优选地表示氢;
[0125]
rc和rd独立地(并且优选地)表示h;
[0126]
l1优选地表示如在-c(ra)(rb)-中所定义的接头基团(对于本发明的主要实施例,该接头基团是必需的);
[0127]
x1可以不存在,但是优选地表示芳香族接头基团(对于本发明的主要实施例,该接头基团当存在时必须是碳环芳香族接头基团);
[0128]
xc(其附接至xa)不存在,或者当xa表示ch时,xc也可以表示-o-;
[0129]
xd不存在,或者当n1表示2或当xc不存在,xa表示c(rc)并且n1表示1时,xd也可以表示-o-;
[0130]
xe和xf独立地表示不存在,或可以独立地表示-o-,其条件是前述的氧原子不直接附接至或α不直接附接至另一个杂原子;
[0131]
当xc和/或xd表示-o-、-nh-或-s-时,应当理解的是这样的杂原子可以不直接附接至(或α不直接附接至)另一个杂原子。
[0132]
本发明的更优选的化合物包括这些,其中:
[0133]
r1表示氢;
[0134]
ra和rb独立地表示氢;
[0135]
l1表示-ch
2-;
[0136]
当x1表示芳香族接头基团(其中附接点可以是经由环系统的任何原子)时,该芳香
族基团可以是碳环或杂环的,从而形成例如苯基、5元或6元单环杂芳基或二环芳香族基团(例如8或10元芳香族基团,其由两个彼此稠合的独立的环组成,其中每个环为5或6元,从而形成6,6-、5,6-或5,5-稠合的二环),因此包括诸如苯基、萘基(包括完全芳香族萘基和1,2,3,4-四氢萘基)等的基团,从而形成例如尤其是:
[0137]-亚苯基-(尤其是1,4-亚苯基),例如:
[0138][0139]-亚萘基,例如:
[0140][0141]-喹啉基(例如2-喹啉基),例如:
[0142][0143]
x1可以表示的此类接头基团(例如亚苯基)可以任选地被取代(例如被一个或多个选自氟、ch3、cf3、-och3和-ocf3的取代基取代)。在一个实施例中,x1可以表示的此类接头基团是未取代的。
[0144]
在本发明的一个实施例(例如上面提到的主要实施例)中,适用以下内容:
[0145]
x1表示任选的碳环芳香族接头基团,即它可以存在或可以不存在;
[0146]
当x1存在时,它表示碳环芳香族接头基团例如苯基或二环(碳环)芳香族接头基团(其中该二环中的至少一个环是芳香族的),例如使得该二环由两个彼此稠合的独立的环组成,其中每个环是5或6元的,从而形成6,6-、5,6-或5,5-稠合的二环),因此包括诸如苯基、萘基(包括完全芳香族萘基和1,2,3,4-四氢萘基)等的基团,从而形成例如尤其是:
[0147]-亚苯基-(尤其是1,4-亚苯基),例如:
[0148][0149]-亚萘基,例如:
[0150][0151]
在本发明的一个方面中,x1,即芳香族接头基团(在一个实施例中,碳环芳香族接头基团,如上面定义的一个)是存在的。
[0152]
螺环部分,即包含组合的xa和xb的环可以如以下所表示:
[0153][0154]
可以提及的其他螺环部分包括以下:
[0155][0156]
因此,可能优选的是:
[0157]
xa表示n或c(rc)(例如ch);
[0158]
xb表示n、o、c(rc)(例如ch)或c=o;
[0159]
xa和xb中的至少一个表示n,并且另一个表示c(rc)、n或(在xb的情况下)o;
[0160]
优选xa和xb均不表示c(rc);
[0161]
xc不存在或表示-o-;
[0162]
xd不存在;
[0163]
xe不存在;
[0164]
xf不存在;
[0165]
xg、xh、xi和xj独立地表示不存在;
[0166]
n1表示0、1或2;
[0167]
n2表示1或2;
[0168]
n3表示1或2;
[0169]
n4表示1或2;
[0170]
l2可以表示氢、卤素(例如氟)、-orf、或芳香族基团(任选地被一个或两个(例如一个)取代基取代(该取代基选自-oc
1-6
烷基(其本身任选地被一个或多个氟原子取代)或-sf5),或者可替代地被卤素(例如氟)取代);
[0171]
更具体地,l2可以表示氢、卤素(例如氟)、-oh、苯基(任选地被-ocf3、-sf5取代和/或可替代地被-och3或氟取代;在另一个实施例中,可以提到的其他取代基包括-scf3)、吡啶基(例如3-吡啶基(优选地是未取代的)、或可替代地是2-或4-吡啶基(也优选地是未取代的))、三唑基或噻唑基;
[0172]
可替代地,可以提到的其他l2基团包括-orf,例如其中rf表示c
1-6
烷基(例如甲基、-ch3)或任选地被c
1-3
烷基(其本身任选地被一个或多个氟原子取代,从而形成像-cf3基团)取代的芳基(例如苯基)或者l2可以表示-c(o)-rg,其中rg表示氢或c
1-3
烷基(例如甲基;任选地被氟取代,从而形成例如-cf3基团)或苯基(优选未取代的);因此l2也可以表示-c(o)h、-c(o)ch3、-c(o)cf3、-c(o)-苯基、-och3或-o-苯基(即苯氧基),后一个基团可以被-cf3部分取代(或者l2和rd可以连接到一起以形成环状基团)。在另一个实施例中,可以另外提到的(例如当附接至氮时,当xb是n时)又其他的l2基团包括任选地被一个或多个氟原子取代的-s(o)
2-c
1-6
烷基基团(例如形成-s(o)2cf3)。
[0173]
在另一个实施例中,xb也可以表示s、s(o),或者在优选的实施例中表示s(o)2。
[0174]
进一步优选的是:
[0175]
q1表示-ch
2-、-ch
2-ch
2-、-o-ch
2-或
“‑”
(即,在后一种情况下,n1=0,xc不存在并且xd不存在);
[0176]
q2表示-ch
2-或-ch
2-ch
2-;
[0177]
q3表示-ch
2-或-ch
2-ch
2-;
[0178]
q4表示-ch
2-或-ch
2-ch
2-。
[0179]
优选地,本发明的化合物包含:
[0180]
环a,其是包含至少1至3个(例如1个或2个)杂原子的,优选地包含至少一个氮原子的芳香族环;
[0181]
环b更优选地也是优选地含有至少一个氮原子的芳香族环(例如5或尤其是6元芳香族环)。
[0182]
优选地,本发明化合物的环a如以下所表示:
[0183][0184]
其他优选的环a部分包括:
[0185][0186]
可以提及的单环杂芳基包括含有1至4个杂原子(优选选自氮、氧和硫)的5或6元环。优选地,本发明化合物的环b如以下所表示:
[0187][0188]
其中“sub”可以是在碳原子上或在可能的情况下在杂原子上(例如在nh上,因此取代h)的相关的任选取代基(或在可能的情况下,可以是多于一个相关的取代基)。
[0189]
其他优选的“环b”部分包括:
[0190]
[0191]
优选的环b上的取代基(当存在时;例如此类任选的取代基可以不存在或可以是一个)包括c
1-3
烷基(例如甲基)或卤素(例如溴或更优选地,氯)。环b上的其他优选的取代基包括-oc
1-6
烷基(例如-oc
1-3
烷基,例如-och3)。
[0192]
环a上优选的取代基(当存在时;优选地有一个或两个取代基)包括c
1-3
烷基(像甲基或乙基)。当l2表示芳香族基团(例如苯基或吡啶基)并且此类基团是取代的时,优选的取代基包括卤素并且尤其是-oc
1-3
烷基(例如-o-甲基),其中后者是被氟取代的,从而形成例如-ocf3基团。
[0193]
组合的环系统,即环a和环b可以如以下所表示:
[0194][0195][0196]
其中“sub”表示在所述二环上(即在环a和/或在环b上)的一个或多个可能的取代基,并且“sub”表示在所述二环的n原子上的可能的任选的取代基(在此上下文中未取代的意指“nh”)。
[0197]
可以提及的其他组合的环a和环b系统包括以下:
[0198][0199][0200]
当环a通过5元a环的“中心”原子附接至氨基部分时,可以提及的组合的环a和环b系统包括以下:
[0201][0202]
具有式(ia)的以下化合物是优选的:
[0203][0204]
其中
[0205]
整数是如在上文中所定义的,并且其中优选地:
[0206]
n1、n2、n3和n4独立地表示1;
[0207]
xa和xb中的至少一个表示n并且另一个表示ch或n。
[0208]
本发明的(例如在上文中)提到的某些化合物用于在结核病的治疗中使用。在此处提及的此类化合物中的某些本身也可以是新颖的。并且在此处提及的此类化合物中的某些
作为药剂/药物可以是新颖的(或作为药物组合物/配制品的组分是新颖的)。因此,在本发明的另一些方面中,提供了下列化合物本身或下列用作药物/药剂的化合物(在后一种情况下,此类化合物可以是药物组合物/配制品的组分):
[0209]
(i)如下所描绘的具有式(ib)的化合物:
[0210][0211]
其中
[0212]
整数是如在上文中所定义的,并且其中优选地:
[0213]
n1、n2、n3和n4独立地表示1;
[0214]
xa和xb中的至少一个表示n并且另一个表示ch或n;
[0215]
(ii)如在上文中所定义的具有式(ia)的化合物,并且其中:
[0216]
l1表示-ch
2-;
[0217]
x1不存在;
[0218]
xa和xb中的至少一个表示n,并且另一个表示c(rc)、n或(在xb的情况下)o;
[0219]
包含xa和xb的螺环3至6元环附接至4至6元环;
[0220]
在一方面,l2表示如在此处所定义的任选地取代的芳香族基团(如在此处所定义的),和/或在另一方面,l2表示-orf,其中rf表示如在此处所定义的任选地取代的芳基基团(如在此处所定义的);
[0221]
当l2表示(任选地取代的)芳香族基团时,它可以是苯基或5或6元杂环基团(例如包含至少一个氮原子,从而形成吡啶基、噻唑基或三唑基环;在主要实施例中,该杂环基团是吡啶基),其中任选的取代基是如在此处所定义的;
[0222]
芳香族l2基团上的任选的取代基选自卤素、c
1-6
烷基、-cf3、-oc
1-6
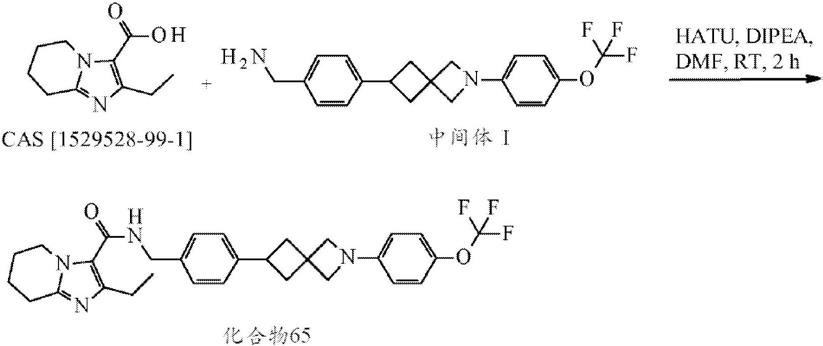
烷基和-ocf3;
[0223]
当rf表示芳基基团时,它优选地是任选地被c
1-3
烷基(其本身任选地被氟取代)取代的苯基);
[0224]
环a和环b一起表示含有至少一个氮原子(并且在主要的实施例中,含有两个环共有的至少一个氮原子)的8或9元二环(环a是5元环并且环b可以是5或6元环,其中两个环均优选地是芳香族的);
[0225]
环a和环b上的任选的取代基是卤素、c
1-3
烷基和oc
1-3
烷基;
[0226]
(iii)如在上文中所定义的具有式(ia)的化合物,并且其中:
[0227]
l1表示-ch
2-;
[0228]
x1表示碳环芳香族接头基团;
[0229]
当x1表示碳环接头基团时,它表示例如亚苯基(例如1,4-亚苯基):
[0230][0231]
xa和xb中的至少一个表示n,并且另一个表示c(rc)、n或(在xb的情况下)o;
[0232]
包含xa和xb的螺环3至6元环附接至4至6元环;
[0233]
在一方面,l2表示如在此处所定义的任选地取代的芳香族基团(如在此处所定义的),和/或在另一方面,l2表示-orf,其中rf表示如在此处所定义的任选地取代的芳基基团(如在此处所定义的);
[0234]
当l2表示(任选地取代的)芳香族基团时,它可以是苯基或5或6元杂环基团(例如包含至少一个氮原子,从而形成吡啶基、噻唑基或三唑基环;在主要实施例中,该杂环基团是吡啶基),其中任选的取代基是如在此处所定义的;
[0235]
在芳香族l2基团上的任选的取代基选自卤素、c
1-6
烷基、-cf3、-oc
1-6
烷基和-ocf3;
[0236]
当rf表示芳基基团时,它优选地是任选地被c
1-3
烷基(其本身任选地被氟取代)取代的苯基);
[0237]
环a和环b一起表示含有至少一个氮原子(并且在主要的实施例中,含有两个环共有的至少一个氮原子)的8或9元二环(环a是5元环并且环b可以是5或6元环,其中两个环均优选地是芳香族的);
[0238]
环a和环b上的任选的取代基是卤素、c
1-3
烷基和-oc
1-3
烷基;
[0239]
(iv)如在上文中所定义的化合物(例如在上述的(i)、(ii)或(iii)中),并且进一步地在其中:
[0240]
q1表示-ch
2-、-ch
2-ch
2-、-o-ch
2-或
“‑”
(即,在后一种情况下,n1=0,xc不存在并且xd不存在);
[0241]
q2表示-ch
2-或-ch
2-ch
2-;
[0242]
q3表示-ch
2-或-ch
2-ch
2-;
[0243]
q4表示-ch
2-或-ch
2-ch
2-;
[0244]
(v)如在上文中所定义的化合物(例如在上述的(i)、(ii)、(iii)或(iv)中),并且进一步地在其中包含xa和xb的环是如在此处定义的所表示的或更具体地是如下:
[0245][0246]
(或上述表示中的任一项);和/或
[0247]
(vi)如在上文中所定义的化合物(例如在上述(i)、(ii)、(iii)、(iv)或(v)中),并且进一步地在其中环a和环b二环是如在此处定义的所表示的或更具体地是如下:
[0248][0249]
(或上述表示中的任一项)。
[0250]
药理学
[0251]
根据本发明的化合物出人意料地显示适于治疗细菌感染,包括分枝杆菌感染,特别是由病原性分枝杆菌,例如结核分枝杆菌(包括其潜伏性和耐药形式)引起的那些疾病。本发明因此还涉及如在上文中所定义的本发明的化合物用作一种药品,特别是用作用于治疗细菌感染(包括分枝杆菌感染)的药品。
[0252]
本发明的这些化合物可以通过干扰结核分枝杆菌中的atp合酶来起作用,其中细胞色素bc1活性的抑制是主要作用方式。细胞色素bc1是atp合成所必需的电子传递链的必要成分。
[0253]
此外,如下文中描述,本发明还涉及本发明的化合物连同任何其药物组合物用于制造用于治疗细菌感染(包括分枝杆菌感染)的药剂的用途。
[0254]
因此,在另一个方面中,本发明提供了一种治疗患有细菌感染(包括分枝杆菌感染)或处于此风险的患者的方法,该方法包括向该患者给予治疗有效量的根据本发明的化合物或药物组合物。
[0255]
本发明的化合物还显示抗耐受性细菌菌株的活性。
[0256]
无论何时在上文或下文中使用,这些化合物可以治疗细菌感染意指这些化合物可以治疗被一种或多种细菌菌株的感染。
[0257]
本发明还涉及一种组合物,该组合物包括药学上可接受的载体以及作为活性成分的、治疗有效量的根据本发明的化合物。根据本发明的这些化合物可以被配制为用于给药目的的不同药物形式。作为适当的组合物,可以提及通常用于全身性给予药物的所有组合物。为了制备本发明的药物组合物,将作为活性成分的有效量的具体化合物(任选地呈加成盐形式)与药学上可接受的载体组合在紧密混合物中,该载体取决于用于给予所希望的制剂的形式可以采用多种形式。所希望地是,这些药物组合物处于单位剂型,具体地是适用于经口给予或通过注射剂给予的单位剂型。例如,在制备处于口服剂型的组合物中,可使用任何常见药物介质,在口服液体制剂(如悬浮液、糖浆剂、酏剂、乳液以及溶液)的情况下,例如像水、二醇类、油类、醇类等;或者在粉剂、丸剂、胶囊剂和片剂的情况中,固体载体诸如淀粉、糖、高岭土、稀释剂、润滑剂、粘合剂、崩解剂以及类似物。因为其容易给予,片剂和胶囊
剂表示了最有利的口服单位剂型,在该情况下显然使用固体药物载体。对于肠胃外组合物来说,载体通常将包括至少呈大部分的无菌水,但也可以包括其他成分例如以辅助溶解性。可以制备例如可注射溶液,其中载体包括生理盐水溶液、葡萄糖溶液或生理盐水与葡萄糖溶液的混合物。还可以制备可注射悬浮液,在该情况下,可以使用适当液体载体、悬浮剂等。还包括旨在使用前不久转化为液体形式制剂的固体形式制剂。
[0258]
取决于给予的模式,该药物组合物将优选地包括按重量计从0.05%至99%,更优选地按重量计从0.1%至70%,甚至更优选地按重量计从0.1%至50%的这种或这些活性成分,以及按重量计从1%至99.95%,更优选地按重量计从30%至99.9%,甚至更优选地按重量计从50%至99.9%的一种药学上可接受的载体,所有的百分数都基于组合物的总重量。
[0259]
该药物组合物另外可以包含本领域已知的不同其他成分,例如,润滑剂、稳定剂、缓冲剂、乳化剂、粘度调节剂、表面活化剂、防腐剂、香味剂或着色剂。
[0260]
为了便于给药和剂量的均一性,将上述药物组合物配制成单位剂型是特别有利的。如在此使用的单位剂型指的是适合作为单位剂量的物理离散单位,各单位含有预定量的活性成分,该预定量的活性成分经计算与所需药物载体相结合而产生所希望的治疗效果。此类单位剂型的实例是片剂(包括刻痕或包衣的片剂)、胶囊剂、丸剂、粉末包(powder packet)、糯米纸囊剂(wafer)、栓剂、可注射溶液或悬浮液以及类似剂型,及其分离的多剂量形式(multiples)。当然,根据本发明的化合物的每日剂量将随着所采用的化合物、给予模式、所希望的治疗以及所针对的分枝杆菌疾病而变化。然而,一般而言,当根据本发明的化合物以不超过1克(例如,在从10至50mg/kg体重的范围内)的每日剂量给予时,将获得令人满意的结果。
[0261]
考虑到具有式(ia)或化学式(ib)的化合物针对细菌感染是有活性的事实,本发明化合物可以与其他抗细菌剂组合以便有效地对抗细菌感染。
[0262]
因此,本发明还涉及(a)根据本发明的化合物,以及(b)一种或多种其他抗细菌剂的组合。
[0263]
本发明还涉及(a)根据本发明的化合物,以及(b)一种或多种其他抗细菌剂的组合,该组合用作药品。
[0264]
本发明还涉及如上文直接定义的组合或药物组合物用于治疗一种细菌感染的用途。
[0265]
本发明还包括药物组合物,该组合物包括药学上可接受的载体以及作为活性成分的治疗有效量的(a)根据本发明的化合物,以及(b)一种或多种其他抗细菌剂。
[0266]
当作为一个组合给出时,本领域的普通技术人员可以确定(a)根据本发明的化合物以及(b)一种或多种其他抗细菌剂的重量比。如本领域的普通技术人员所熟知的,所述比率以及精确的剂量以及给予的频率取决于根据本发明的具体化合物以及所使用的一种或多种其他抗细菌剂、正治疗的具体病症、正治疗的病症的严重性、具体患者的年龄、体重、性别、饮食、给予的时间以及总体身体健康状况、给予模式连同个体可以服用的其他药物。此外,显然该有效的每日用量可以降低或提高,这取决于所治疗的受试者的响应和/或取决于给出本发明化合物处方的医生的评估。本发明的化合物与另一种抗细菌剂的具体重量比可以在从1/10到10/1、更尤其从1/5到5/1、甚至更尤其从1/3到3/1的范围内。
[0267]
根据本发明的这些化合物以及该一种或多种其他抗细菌剂可以组合在一个单一
制剂中或者它们可以被配置为分开的制剂,这样使得它们可以同时地、分开地或顺序地给予。因此,本发明还涉及一种产品,该产品包含(a)根据本发明的化合物,以及(b)一种或多种其他抗细菌剂,以作为一个组合的制剂,用于在细菌感染的治疗中同时地、分开地或顺序地使用。
[0268]
可以与本发明的化合物组合的其他抗细菌剂是例如本领域已知的抗细菌剂。例如,本发明的化合物可以与已知干扰结核分枝杆菌的呼吸链的抗菌剂组合,所述抗菌剂包括例如atp合酶的直接抑制剂(例如贝达喹啉、贝达喹啉富马酸酯或可披露于现有技术中的任何其他的化合物,例如wo2004/011436中披露的化合物)、ndh2抑制剂(例如氯法齐明)和细胞色素bd抑制剂。可与本发明组合物组合的另外的分枝杆菌剂是例如利福平(=雷发平);异烟肼;吡嗪酰胺;阿米卡星;乙硫异烟胺;乙胺丁醇;链霉素;对氨基水杨酸;环丝氨酸;卷曲霉素;卡那霉素;氨硫脲;pa-824;迪拉马尼(delamanid);喹诺酮类/氟喹诺酮类,如莫西沙星、加替沙星、氧氟沙星、环丙沙星、司帕沙星;大环内酯,例如,如克拉霉素、与克拉维酸组合的阿莫西林;利福霉素;利福布汀;利福喷汀;以及目前正在进行研发的其他分枝杆菌剂(但是可能仍然未出现在市场上;参见例如http://www.newtbdrugs.org/pipeline.php)。
[0269]
通用制备
[0270]
根据本发明的这些化合物通常可以通过一系列步骤进行制备,其中的每个步骤可以是熟练人员已知的或在此处所描述的。
[0271]
实验部分
[0272]
具有式i的化合物可根据下文实例中所用的技术(以及本领域技术人员已知的那些方法)来制备,例如通过使用以下技术来制备。
[0273]
具有式(i)或(ia)的化合物,其中xb表示n,可以通过如下步骤来制备:
[0274]
(i)使具有式(ii)的化合物,
[0275][0276]
其中整数是如在上文中所定义的,和具有式(iii)的化合物反应,
[0277]
lg
1-l2ꢀꢀꢀ
(iii)
[0278]
其中l2是如在上文中所定义的(例如当l2不是氢、卤素或连接至o或s上时),并且lg1是合适的离去基团例如氯、溴、碘或磺酸酯基团,该反应可能要求特定的条件(例如像在此处所述的亲核性芳香族取代反应条件);
[0279]
(ii)使具有式(iv)的化合物,
[0280][0281]
其中整数是如上文所定义的,或其合适的衍生物,例如羧酸酯衍生物,与具有式(v)的化合物
[0282][0283]
其中整数是如在上文中所定义的,在酰胺偶联反应条件下,例如在合适的偶联剂(例如1,1
’‑
羰基二咪唑、n,n
’‑
二环己基碳二亚胺、1-(3-二甲基氨丙基)-3-乙基碳二亚胺(或其盐酸盐)或n,n
’‑
二琥珀酰亚胺基碳酸酯)的存在下,任选地在合适的碱(例如氢化钠、碳酸氢钠、碳酸钾、吡啶、三乙胺、二甲氨基吡啶、二异丙基胺、氢氧化钠、叔丁醇钾和/或二异丙氨基锂(或其变体)和适宜的溶剂(例如四氢呋喃、吡啶、甲苯、二氯甲烷、氯仿、乙腈、二甲基甲酰胺、三氟甲基苯、二噁烷或三乙胺)的存在下反应。可替代地,具有式(iv)的化合物的羧酸基团可以首先在标准条件下被转化为相应的酰氯(例如在pocl3、pcl5、socl2或草酰氯的存在下),然后例如在与上述那些相似的条件下,该酰氯与具有式(v)的化合物反应;
[0284]
(iii)使具有式(vi)的化合物,
[0285][0286]
其中整数是如在上文中所定义的,并且lg2表示合适的离去基团,例如碘、溴、氯或磺酸酯基团(例如可以用于偶联的一种基团),与一种具有式(vi)的化合物,
[0287][0288]
其中整数是如在上文中所定义的,在标准条件下,例如任选地在适宜的金属催化剂(或其一种盐或络合物)例如pd(dba)2、pd(oac)2、cu、cu(oac)2、cui、nicl2等等、以及任选的添加剂例如ph3p、x-phos等等的存在下,在适宜的碱(例如t-buona、等等)的存在下,在合适的溶剂(例如二噁烷等等)中,在本领域技术人员已知的反应条件下偶联;
[0289]
(iv)使具有式(viii)的化合物,
[0290][0291]
其中整数是如在上文中所定义的,并且lg3表示如上文所述的相对于lg2的合适的离去基团(并且可以具体地表示氯、溴或碘),与一种具有式(ix)的化合物偶联,
[0292]
lg
4-l2ꢀꢀꢀ
(ix)
[0293]
其中l2是如在上文中所定义的(例如,当l2不是氢、卤素或连接至o或s时),并且lg4是合适的基团例如-b(oh)2、-b(or
wx
)2或-sn(r
wwx
)3,其中每个r
wx
独立地表示c
1-6
烷基基团,或者在-b(or
wx
)2的情况下,各r
wx
基团可以连接到一起以形成4至6元的环状基团,从而形成像频哪醇并硼酸酯基团(或者lg4可以表示碘、溴或氯,其条件是lg3和lg4是相互兼容的),并且其中该反应可以在一种合适的催化剂系统,像金属(或其一种盐或络合物)例如pd、cui、pd/c、pdcl2、pd(oac)2、pd(ph3p)2cl2、pd(ph3p)4、pd2(dba)3和/或nicl2(等等)和一种配体例如pdcl2(dppf).dcm、t-bu3p、(c6h
11
)3p、ph3p等等的存在下,在合适的溶剂中并且在本领域技术人员已知的反应条件下进行。
[0294]
显然在前述和以下反应中,这些反应产物可以从该反应介质中分离出,并且,如果需要的话,根据本领域中通常已知的方法学(诸如,萃取、结晶和层析法)进行进一步纯化。更加显然,以多于一种对映异构体的形式存在的反应产物可以通过已知的技术(具体地是制备型层析,诸如制备型hplc、手性层析)从它们的混合物中分离。还可以通过超临界流体层析法(scf)获得单独的非对映异构体或单独的对映异构体。
[0295]
这些起始材料以及中间体是可商购的化合物或根据本领域中通常已知的常规反应程序可以制备的化合物。
[0296]
化合物1的合成
[0297][0298]
中间体a的制备
[0299]
在-70℃,在n2流下,将lihmds(50ml,1m在thf中)添加到n-叔-丁氧基羰基-4-哌啶酮(cas[79099-07-3],8.86g,50.0mmol)在thf(180ml)中的混合物中。将混合物搅拌10分钟。在-70℃下,将二乙基氰甲基膦酸酯(9g,45.2mmol)添加至混合物中。将该混合物搅拌1小时。将混合物用nh4cl溶液淬灭,用乙酸乙酯萃取,用盐水洗涤,经mgso4干燥并过滤。将滤液浓缩以给出10.0g,90.0%的a。
[0300]
中间体b的制备
[0301]
将me3soi(10.9g,49.5mmol)缓慢地添加至t-buok(5.55g,49.5mmol)在dmso(60ml)中的溶液中。将该混合物搅拌1.5小时。将a(10.0g,45.0mmol)在dmso(80ml)中的溶液添加至混合物中。将混合物在45℃下搅拌24小时。将饱和nh4cl溶液添加至混合物中并且搅拌0.5小时。将该混合物用乙酸乙酯进行萃取。将有机层用盐水洗涤,经mgso4干燥并过滤。将滤液浓缩以给出10.0g,93%的b。
[0302]
中间体c的制备
[0303]
向b(460mg,1.95mmol)在meoh(10ml)中的溶液中添加cocl2·
6h2o(463mg,1.95mmol)。将混合物在-10℃下搅拌10分钟。在混合物上分批添加nabh4(368mg,9.74mmol)。然后将混合物再搅拌1小时。添加1mhcl水溶液并且将固体溶解。将水相用nh3·
h2o水溶液碱化至ph=9,并用乙酸乙酯萃取。将合并的有机层经na2so4干燥并在真空中浓缩。将残余物用乙酸乙酯中的草酸溶液研磨并过滤以给出白色固体。将固体用1n naoh水溶液碱化并用二氯甲烷萃取。将合并的有机层经na2so4干燥并在真空中浓缩以给出120mg,26%的c。
[0304]
制备中间体d
[0305]
将hobt(55.1mg,0.408mmol)、6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],91.7mg,0.408mmol)、diea(105mg,0.816mmol)和edci
·
hcl(117mg,
0.612mmol)添加至c(100mg,0.416mmol)在dmf(10ml)中的搅拌溶液中。将混合物搅拌并在60℃下加热16小时。将混合物浓缩。将该残余物溶解在乙酸乙酯中。有机层用h2o洗涤,经mgso4干燥并过滤。将滤液浓缩以给出100mg,51%的d。
[0306]
制备中间体e
[0307]
在0℃下,将tfa(5ml)添加至d(90mg,0.201mmol)在ch2cl2(5ml)中的混合物中。将该混合物在室温下搅拌5小时。将混合物在真空下浓缩。将残余物溶于ch2cl2中,并且用nahco3溶液将混合物调节至ph=7。将有机层分离并浓缩。将该粗产物经硅胶柱层析法(洗脱液:乙酸乙酯/石油醚为从0至1)进行纯化。将产物级分收集并浓缩以给出70mg,90%的e。
[0308]
化合物1的制备
[0309]
将e(20mg,0.058mmol)、1-碘-4-(三氟甲氧基)苯(cas[103962-05-6],16.7mg,0.058mmol)、pd(dba)2(3.34mg,0.006mmol)、xphos(4.57mg,0.009mmol)和t-buona(22.3mg,0.232mmol)在1,4-二噁烷(5ml)中的溶液在110℃下在微波下在n2下辐射1小时。将混合物在真空下浓缩。将残余物经gemini通过高效液相层析法进行纯化(洗脱液:nh3水/乙腈30/70至70/30)。收集所需级分并浓缩,以给出化合物1,19.3mg,64%。
[0310]1h nmr(400mhz,cdcl3)δppm 9.47(s,1h)7.54(d,j=9.29hz,1h)7.30(dd,j=9.41,1.83hz,1h)7.10(d,j=8.80hz,2h)6.91(d,j=9.05hz,2h)5.87(br.s.,1h)3.51-3.60(m,2h)3.30-3.42(m,2h)3.08-3.17(m,2h)3.02(q,j=7.58hz,2h)1.86-1.94(m,1h)1.73-1.82(m,1h)1.64-1.69(m,1h)1.43(t,j=7.58hz,3h)1.36(d,j=13.45hz,1h)1.01-1.10(m,1h)0.70(dd,j=8.44,4.77hz,1h)0.38(t,j=4.89hz,1h)
[0311]
化合物2的合成
[0312][0313]
中间体f的制备
[0314]
在氮气流下,在25℃下,将中间体r(364mg,2.47mmol)、反式-2-氨基-环己醇(8.5mg,0.248mmol)和碘化镍(38.7mg,0.124mmol)在i-proh(4ml)中的混合物搅拌30分钟。添加nahmds(2.48ml,1m在thf中),并且在氮气流下将混合物搅拌10分钟。添加4-氰基苯硼酸(cas[126747-14-6],400mg,1.24mmol)在i-proh(4ml)中的溶液并且将混合物在60℃下在微波下搅拌1小时,在90℃下搅拌1小时并且在120℃下搅拌4小时。将混合物用二氯甲烷(50ml)稀释,用水(2
×
50ml)和盐水(20ml)洗涤。将有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯5/1)进行纯化以给出中间体f(300mg,产率:37%)。
[0315]
中间体g的制备
[0316]
将中间体f(300mg,1.01mmol)在甲酸(5ml)中的混合物在室温下搅拌12小时。将混合物浓缩并将ch2cl2(30ml)添加至混合物中。用na2co3溶液(20ml)洗涤混合物。将有机层分离,经na2so4干燥并过滤。将滤液浓缩以给出中间体g(150mg,产率:64%)。
[0317]
中间体h的制备
[0318]
在n2下,将中间体g(100mg,0.504mmol)、1-碘-4-(三氟甲氧基)苯(cas[103962-05-6],145mg,0.504mmol)、x-phos(28.8mg,0.06mmol)、pd(dba)2(17.4mg,0.03mmol)和t-buona(194mg,2.02mmol)在二噁烷(4ml)中的溶液在100℃下在微波下辐射1小时。将混合物浓缩。将该粗产物经硅胶柱层析法(洗脱液:乙酸乙酯/石油醚为从0至1/l)进行纯化。将这些所希望的级分进行收集并且进行浓缩以给出中间体h(100mg,产率:55%)。
[0319]
中间体i的制备
[0320]
将中间体h(70.0mg,0.195mmol)在nh3·
meoh(7m,在甲醇溶液中,20ml)中的混合物用拉尼镍(7mg)作为催化剂在25℃下氢化(15psi)16小时。在吸收了h2之后,将催化剂过滤出,并且将滤液进行浓缩以给出中间体i(50.0mg,产率:71%)。
[0321]
化合物2的制备
[0322]
在25℃下,将6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],22.5mg,0.100mmol)、hatu(49.4mg,0.130mmol)、diea(33.6mg,0.260mmol)在ch2cl2(20ml)中的溶液搅拌30分钟。将中间体i(40.0mg,0.110mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:0.05%氨在水/甲醇中20/80至5/95)。将这些所希望的级分进行收集并且进行浓缩以给出化合物2(9.80mg,产率:17%)。
[0323]
1h nmr(400mhz,cdcl3)δ=ppm 9.54(s,1h)7.55(d,j=9.26hz,1h)7.27-7.37(m,3h)7.22(d,j=7.94hz,2h)7.00-7.10(m,2h)6.40(d,j=8.82hz,2h)6.11(br.s.,1h)4.68(d,j=5.73hz,2h)4.01(s,2h)3.80(s,2h)3.48(q,j=8.93hz,1h)2.98(q,j=7.50hz,2h)2.59-2.71(m,2h)2.35(td,j=9.70,2.65hz,2h)1.36-1.47(m,3h)
[0324]
化合物3的合成
[0325][0326]
中间体j的制备
[0327]
将nbs(45.1g,254mmol)和nh4oac(5.33g,69.2mmol)添加至3-氧代戊酸甲酯(cas[30414-53-0],30g,231mmol)在甲基叔-丁基醚(600ml)中的溶液中。将混合物在室温下搅拌48小时。过滤混合物并用h2o洗涤,经na2so4干燥并过滤。将滤液在真空下进行浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯20/1)进行纯化以给出中间体j(20.0g,产率:35%)。
[0328]
中间体k的制备
[0329]
将5-氯-2-吡啶胺(cas[5428-89-7],12.0g,93.0mmol)和中间体j(25.0g,112mmol)在乙醇(60ml)中的溶液回流过夜。将混合物在真空下浓缩。将该残余物溶解在乙酸乙酯(100ml)中。将溶液用水(2
×
100ml)、盐水(100ml)洗涤,经硫酸钠干燥,过滤并在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯3/1)进行纯化以给出中间体k(700mg,产率:3%)。
[0330]
中间体l的制备
[0331]
将中间体k(700mg,2.10mmol)和氢氧化钠(252mg,6.30mmol)在乙醇(2ml)和h2o(2ml)中的混合物在室温下搅拌过夜。添加水(20ml),并将溶液用2m盐酸盐水溶液酸化至ph约为3。将该溶液冻干以给出粗中间体l(2g)。
[0332]
化合物3的制备
[0333]
相应地,从中间体l和中间体i开始以与化合物2相同的方式制备化合物3,产量9.60mg,产率:8%。
[0334]
1h nmr(400mhz,cdcl3)δppm 9.84(d,j=2.51hz,1h)8.57(d,j=2.76hz,1h)7.30-7.35(m,2h)7.22(d,j=8.03hz,2h)7.06(d,j=8.03hz,2h)6.37-6.43(m,2h)6.14-6.20(m,1h)4.68(d,j=5.77hz,2h)4.01(s,2h)3.80(s,2h)3.48(q,j=8.85hz,1h)3.02(q,j=7.53hz,2h)2.61-2.70(m,2h)2.31-2.40(m,2h)1.45(t,j=7.53hz,3h)
[0335]
化合物4的合成
[0336][0337]
中间体m的制备
[0338]
将6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[12161242-18-5],1g,4.45mmol)、4-碘苯甲胺(cas[39959-59-6],1.09g,4.67mmol)、edci
·
hcl(1.28g,6.68mmol)、hobt(0.601g,4.45mmol)和三乙胺(1.24ml,9mmol)在二氯甲烷(8ml)中的溶液搅拌并且在45℃下加热24小时。将溶液冷却至15℃。过滤收集固体,用水和乙腈洗涤,并干燥固体(真空,45℃,1小时)以给出中间体m,1.2g,55%。
[0339]
中间体n的制备
[0340]
在n2下,将叔-丁基2,6-二氮杂螺[3.3]庚烷-2-甲酸酯(cas[1041026-70-3],500mg,2.52mmol)、1-碘-4-(三氟甲氧基)苯(cas[103962-05-6],726mg,2.52mmol)、x-phos(240mg,0.504mmol)、pd(dba)2(145mg,0.252mmol)和t-buona(969mg,10.1mmol)在二噁烷(8ml)中的溶液在110℃下在微波下辐射1小时。将水添加至该混合物并且将该混合物用乙酸乙酯(50mlx2)萃取。将有机层用盐水洗涤,经mgso4干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(洗脱液:乙酸乙酯/己烷为从0至1/5)进行纯化。将所希望的级分收集并浓缩以给出500mg,50%的n。
[0341]
中间体o的制备
[0342]
将n(100mg,0.279mmol)在hcooh(5ml)中的混合物搅拌12小时。将该混合物浓缩并用于下一步而无需进一步纯化。
[0343]
化合物4的制备
[0344]
在n2下,将中间体o(72mg,0.279mmol)、中间体m(123mg,0.279mmol)、x-phos(6.6mg,0.056mmol)、pd(dba)2(16.0mg,0.028mmol)和t-buona(107mg,1.12mmol)在二噁烷(8ml)中的溶液在110℃下在微波下辐射1小时。将混合物浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:氨在水/乙腈中50/50至20/80)。收集所需级分并浓缩,以给出化合物4,35.8mg,22%。
[0345]
1h nmr(400mhz,cdcl3)δppm=9.53(d,j=1.25hz,1h)7.56(d,j=9.79hz,1h)7.31(dd,j=9.54,2.01hz,1h)7.24(s,2h)7.08(d,j=8.53hz,2h)6.49(d,j=8.53hz,2h)6.42(d,j=9.03hz,2h)6.01(br.s.,1h)4.59(d,j=5.27hz,2h)4.04(s,4h)4.02(s,4h)2.96(q,j=7.36hz,2h)1.39(t,j=7.53hz,3h)
[0346]
化合物5的合成
[0347][0348]
中间体p的制备
[0349]
在n2下,将中间体o(100mg,0.387mmol)、4-碘苯基氰(cas[3058-39-7],115mg,0.503mmol)、x-phos(22.0mg,46.2mmol)、pd(dba)2(13.3mg,23.1mmol)和t-buona(149mg,1.55mmol)在二噁烷(5ml)中的溶液在110℃下在微波下辐射1小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:0.05%氨在水/甲醇中30/70至5/95)。将这些所希望的级分进行收集并且进行浓缩以给出中间体p(60.0mg,产率:35%)。
[0350]
中间体q的制备
[0351]
相应地,从中间体p开始以与中间体i相同的方式制备中间体q,产量60.0mg,产率:99%。
[0352]
化合物5的制备
[0353]
在25℃下,将中间体l(28.3mg,0.125mmol)、hatu(61.8mg,0.162mmol)、diea(42.0mg,0.325mmol)在dmf(5ml)中的溶液搅拌30分钟。将中间体q(50.0 mg,0.138mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:0.05%氨在水/甲醇中25/75至5/95)。将这些所希望的级分进行收集并且进行浓缩以给出化合物5(10.3mg,产率:14%)。
[0354]
1h nmr(400mhz,cdcl3)δ=ppm9.84(d,j=2.51hz,1h)8.56(d,j=2.51hz,1h)7.25(d,j=8.53hz,2h)7.08(d,j=8.78hz,2h)6.49(d,j=8.28hz,2h)6.43(d,j=9.03hz,2h)6.06(s,1h)4.59(d,j=5.27hz,2h)4.05(s,4h)4.03(s,4h)2.99(q,j=7.45hz,2h)1.43(t,j=7.53hz,3h)
[0355]
化合物6的合成
[0356][0357]
相应地,化合物6以与化合物5相同的方式从2-乙基-5h,6h,7h,8h-咪唑并[1,2-a]吡啶-3-甲酸cas[1529528-99-1]和中间体q开始制备,产量153.90mg,产率:32%。
[0358]
1h nmr(400mhz,cdcl3)δppm 7.21(d,j=8.28hz,2h)7.08(d,j=8.03hz,2h)6.47(d,j=8.53hz,2h)6.40-6.45(m,2h)5.83(br.s.,1h)4.50(d,j=5.52hz,2h)4.23(t,j=5.77hz,2h)4.04(s,8h)2.86(t,j=6.40hz,2h)2.68(q,j=7.53hz,2h)1.83-2.01(m,4h)1.23(t,j=7.53hz,3h)
[0359]
化合物7的合成
[0360][0361]
中间体r的制备
[0362]
将三苯基膦(1.89g,7.20mmol)、咪唑(735mg,10.8mmol)和碘(1.37g,5.40mmol)添加至叔-丁基6-羟基-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1147557-97-8],768mg,3.60mmol)在甲苯(50ml)中的溶液中。将所得混合物回流1小时。将混合物冷却至25℃,用水(100ml)和盐水(50ml)洗涤。将分离的有机层干燥,过滤并将该滤液在真空下浓缩。将残余物经硅胶柱快速层析法(洗脱液:石油醚/乙酸乙酯1/0至1/1)进行纯化以给出中间体r(1.20g,产率:93%)。
[0363]
中间体s的制备
[0364]
在氮气流下,将4-(三氟甲氧基)苯基硼酸(cas[139301-27-2],510mg,2.48mmol)、反式-2-氨基-环己醇(23.0mg,0.200mmol)和碘化镍(62.5mg,0.200mmol)在异丙醇(4ml)中的混合物在25℃下搅拌30分钟。添加nahmds(2.47ml,1m在thf中,2.47mmol),并且在氮气流下将混合物搅拌10分钟。添加在异丙醇(1ml)中的中间体r(400mg,1.24mmol)并且将混合物在60℃下在微波下搅拌1小时,在90℃下搅拌1小时并在120℃下搅拌5小时。将混合物用二氯甲烷(50ml)稀释,用水(2
×
50ml)和盐水(20ml)洗涤。将有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯5/1)进行纯化以给出中间体s(230mg,产率:52%)。
[0365]
中间体t的制备
[0366]
在0℃下,在氮气气氛中,将中间体s(220mg,0.616mmol)添加至甲酸(5ml)中。将该混合物在25℃下搅拌5小时。将混合物在真空下浓缩。将该残余物溶解在二氯甲烷(20ml)中。将溶液用饱和碳酸钠水溶液(20ml)、盐水(20ml)洗涤,经硫酸钠干燥,过滤并在真空下浓缩,以给出中间体t(150mg,产率:85%)。
[0367]
化合物7的制备
[0368]
在n2气氛下,将中间体t(110mg,0.428mmol)、中间体m(226mg,0.514mmol)、pd(dba)2(14.8mg,0.0260mol)、x-phos(20.4mmol,0.0430mmol)和叔丁醇钠(165mg,1.71mmol)在1,4-二噁烷(5ml)中的溶液在100℃下在微波下辐射1小时。添加乙酸乙酯(30ml)并且将混合物用水(10ml)和盐水(20ml)洗涤。将有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法进行纯化(洗脱液:石油醚/乙酸乙酯i/0至0/1),以给出粗化合物。将其经菲罗门(phenomenex)gemini c18200
×
25mm
×
10μm,通过高效液相层析法(洗脱液:0.5%氨在水/乙腈中80/20至14.5/85.5)进一步纯化。将这些所希望的级分进行收集并且进行冻干以给出化合物7(84.60mg,产率:35%)。
[0369]1h nmr(400mhz,cdcl3)δ=9.52(d,j=1.8hz,1h),7.53(d,j=9.5hz,1h),7.29(dd,j=2.0,9.5hz,1h),7.26-7.18(m,4h),7.18-7.12(m,2h),6.47(d,j=8.5hz,2h),5.99(br.s.,1h),4.58(d,j=5.3hz,2h),4.02(s,2h),3.80(s,2h),3.47(q,j=8.9hz,1h),2.94(q,j=7.5hz,2h),2.70-2.61(m,2h),2.38-2.29(m,2h),1.38(t,j=7.5hz,3h)
[0370]
化合物8的合成
[0371][0372]
中间体u的制备
[0373]
相应地,中间体u以与中间体h相同的方式制备,从中间体t和4-碘苯基氰cas[3058-39-7]开始,产量120mg,产率:40%。
[0374]
中间体v的制备
[0375]
相应地,中间体v以与中间体i相同的方式制备,从中间体u开始,产量120mg,产率:92%。
[0376]
化合物8的制备
[0377]
将中间体v(125mg,0.222mmol)、中间体l(80.5mg,0.222mmol)、hatu(110mg,
0.289mmol)和diea(74.6mg,0.577mmol)在二氯甲烷(10ml)中的混合物在25℃下搅拌2小时。添加二氯甲烷(50ml)并且将混合物用水(50ml)和盐水(50ml)洗涤。将分离的有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法进行纯化(洗脱液:乙酸乙酯)以给出粗产物。将粗产物经gemini通过高效液相层析法以150
×
255μm(洗脱液:0.05%氨在水/乙腈21/79)进一步纯化。将这些所希望的级分进行收集并且进行冻干以给出化合物8(36.6mg,产率:28%)。
[0378]1h nmr(400mhz,cdcl3)δ=9.83(d,j=2.2hz,1h),8.55(d,j=2.2hz,1h),7.25-7.08(m,6h),6.46(d,j=7.9hz,2h),6.06(br.s.,1h),4.58(d,j=5.3hz,2h),4.02(s,2h),3.81(s,2h),3.47(q,j=8.8hz,1h),2.98(q,j=7.5hz,2h),2.73-2.59(m,2h),2.41-2.27(m,2h),1.42(t,j=7.5hz,3h).
[0379]
化合物9的合成
[0380][0381]
中间体w的制备
[0382]
相应地,中间体w以与中间体h相同的方式制备,从中间体aw(120mg,0.693mmol)和4-碘苯基氰(cas[3058-397],238mg,1.04mmol)开始,产出100mg,52%。
[0383]
中间体x的制备
[0384]
相应地,中间体x以与中间体i相同的方式制备,从中间体w(100mg,0.364mmol)开始,产量100mg,94%。
[0385]
化合物9的制备
[0386]
在25℃下,将中间体l(50.0mg,0.222mmol)、hatu(110mg,0.289mmol)、diea(74.6mg,0.577mmol)在dmf(5ml)中的溶液搅拌30分钟。将中间体x(68.0mg,0.244mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:梯度0.05%氨在水/甲醇中从25/75至5/95)。将这些所希望的级分进行收集并且进行浓缩以给出化合物9(34.7mg,产率:31%)。
[0387]
1h nmr(400mhz,cdcl3)δppm 9.82(d,j=2.51hz,1h)8.55(d,j=2.76hz,1h)7.28-7.35(m,2h)7.18-7.23(m,5h)6.41-6.50(m,2h)6.08(t,j=5.02hz,1h)4.58(d,j=5.52hz,2h)4.00-4.04(m,2h)3.77-3.83(m,2h)3.42-3.53(m,1h)2.98(q,j=7.36hz,2h)2.62-2.69(m,2h)2.33-2.40(m,2h)1.37-1.46(m,3h)
[0388]
化合物10的合成
[0389][0390][0391]
中间体y的制备
[0392]
将4-溴苯基硫五氟化物(cas[774-93-6]4g,14.1mmol)、双(频哪醇并)二硼(cas[73183-34-3],4.30g,16.9mmol)、乙酸钾(2.80g,28.5mmol)和pd(dppf)2cl2(0.946g,1.29mmol)在1,4-二噁烷(50ml)中的混合物在100℃下搅拌16个小时。添加乙酸乙酯(200ml)并且将混合物用水(100ml)和盐水(100ml)洗涤。将分离的有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯10/1)进行纯化以给出中间体y(4.60g,产率:89%)。
[0393]
中间体z的制备
[0394]
在0℃下,将高碘酸钠(3.49g,16.3mmol)逐份添加至中间体y(1.80g,5.45mmol)在浓缩的盐酸盐(5ml)和thf(20ml)中的溶液中。将混合物在室温下搅拌3小时。添加乙酸乙酯(50ml)并且将混合物用饱和亚硫酸钠水溶液洗涤(2x20ml)。将分离的有机层用水(20ml)、
盐水(50ml)洗涤,经硫酸钠干燥,过滤并且在真空下浓缩以给出中间体z(1g,产率:72%)。
[0395]
中间体aa的制备
[0396]
在氮气流下,将中间体z(500mg,2.02mmol)、反式-3-氨基-环己醇(11.5mg,0.100mmol)和碘化镍(31.3mg,0.100mmol)在异丙醇(7ml)中的混合物在室温下搅拌30分钟。添加nahhmds(2.02ml,2.02mmol,1m在thf中),并且将混合物在氮气流下搅拌10分钟。添加中间体r(326mg,1.01mmol)在异丙醇(3ml)中的溶液并且将混合物在60℃下在微波下搅拌1小时,在90℃下搅拌1小时并在120℃下搅拌4小时。将混合物用二氯甲烷(50ml)稀释,用水(50ml)和盐水(50ml)洗涤。将有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯5/1)进行纯化以给出中间体aa(170mg,产率:43%)。
[0397]
中间体ab的制备
[0398]
相应地,中间体ab以与中间体g相同的方式制备,从中间体aa(170mg,0.426mmol)开始,产量100mg,78%。
[0399]
中间体ac的制备
[0400]
相应地,中间体ac以与中间体h相同的方式制备,从中间体ab(80.0mg,0.267mmol)和4-碘苯基氰(cas[3058-397],91.6mg,0.4mmol)开始,产量90mg,71%。
[0401]
中间体ad的制备
[0402]
相应地,中间体ad以与中间体i相同的方式制备,从中间体ac(80.0mg,0.200mmol)开始,产量80mg,99%。
[0403]
化合物10的制备
[0404]
将6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],44.5mg,0.198mmol)、中间体ad(80mg,0.198mmol)、hatu(97.9mg,0.257mmol)和diea(76.8mg,0.594mmol)在dmf(4ml)中的混合物在室温下搅拌2小时。经沃特斯(waters)xbridge prep obd c18 150x305μm(洗脱液:0.05%氨在水/甲醇中15/85至5/95)通过高效液相层析法纯化混合物。将这些所希望的级分进行收集并且进行冻干以给出化合物10(36.6mg,产率:28%)。
[0405]1h nmr(400mhz,cdcl3)δ=9.52(s,1h),7.69(d,j=8.4hz,2h),7.54(d,j=9.3hz,1h),7.34-7.27(m,2h),7.23(br.s.,3h),6.47(d,j=7.9hz,2h),5.99(br.s.,1h),4.58(d,j=4.4 hz,2h),4.03(s,2h),3.81(s,2h),3.52(quin,j=8.5hz,1h),2.94(q,j=7.4hz,2h),2.69(t,j=9.5hz,2h),2.36(t,j=10.1hz,2h),1.38(t,j=7.3hz,3h)。
[0406]
化合物11的合成
[0407][0408]
中间体ae的制备
[0409]
在氮气流中,将中间体r(608mg,4.95mmol)、反式-2-氨基-环己醇(57.0mg,0.495mmol)和nii2(77.3mg,0.248mmol)在i-proh(6ml)中的混合物在25℃下搅拌30分钟。添加nahmds(908mg,4.95mmol),并且将混合物在氮气流下搅拌10分钟。添加在i-proh(4ml)中的3-吡啶硼酸(cas[1692-25-7],800mg,2.48mmol)并且将混合物在微波下在60℃下搅拌1小时,在90℃下搅拌1小时并且在120℃下搅拌4小时。将混合物用二氯甲烷(50ml)稀释,用水(50ml)和盐水(20ml)洗涤。将有机层经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯1)进行纯化以给出中间体ae(250mg,产率:37%)。
[0410]
中间体af的制备
[0411]
相应地,中间体af以与中间体g相同的方式制备,从中间体ae(200mg,0.729mmol)开始,产量120mg,94%。
[0412]
中间体ag的制备
[0413]
相应地,中间体ag以与中间体ag相同的方式制备,从中间体af(80.0mg,0.459mmol)和4-碘苯基氰(cas[3058-39-7],158mg,0.688mmol)开始,产量80.0mg,63%。
[0414]
中间体ah的制备
[0415]
相应地,中间体ah以与中间体i相同的方式制备,从中间体ag(70.0mg,0.254mmol)开始,产量70.0mg,99%。
[0416]
化合物11的制备
[0417]
在25℃下,将中间体l(51.4mg,0.228mmol)、hatu(113mg,0.296mmol)、diea(76.6mg,0.593mmol)在dmf(10ml)中的溶液搅拌30分钟。将中间体ah(70.0mg,0.251mmol)
添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:梯度0.05%氨在水/甲醇中从30/70至5/95)。将这些所希望的级分进行收集并且进行浓缩以给出化合物11(10.5mg,产率:9%)。
[0418]
1h nmr(400mhz,cdcl3)δppm 9.83(d,j=2.51hz,1h)8.55(d,j=2.76hz,1h)8.42-8.49(m,2h)7.53(d,j=7.78hz,1h)7.23(d,j=8.53hz,3h)6.47(d,j=8.53hz,2h)6.06(br.s.,1h)4.58(d,j=5.27hz,2h)4.04(s,2h)3.83(s,2h)3.50(q,j=8.72hz,1h)2.99(q,j=7.53hz,2h)2.65-2.74(m,2h)2.33-2.43(m,2h)1.42(t,j=7.53hz,3h)
[0419]
化合物12和化合物13的合成
[0420][0421]
将6-乙基-2-甲基咪唑并[2,1-b]噻唑-5-甲酸(cas[1131613-58-5],40mg,0.19mmol)、(4-{2-氮杂螺[3.3]庚-2-基}苯基)甲胺(cas[1508720-12-4],46mg,0.23mmol)、edci
·
hcl(29mg,0.15mmol)、hobt(26mg,0.19mmol)和dipea(0.033ml,0.19mmol)在二氯甲烷(1.3ml)和thf(1.3ml)中的溶液在室温下搅拌18小时。将混合物用硅胶扩展并且在真空中蒸发。通过制备型lc(常规sioh 30μm,12g因特奇美拉(interehim),干法填充,流动相梯度:庚烷/etoac从70/30至50/50)纯化残余物,蒸发后给出41mg呈白色固体的化合物12(55%)。
[0422]1h nmr(500mhz,dmso-d6)δppm1.19(t,j=7.4hz,2h)1.70-1.86(m,2h)2.15(t,j=7.6hz,4h)2.42(d,j=1.3hz,3h)2.84(q,j=7.6hz,2h)3.72(s,4h)4.34(d,j=6.0hz,2h)6.36(d,j=8.5hz,2h)7.14(d,j=8.5hz,2h)7.88(d,j=1.3hz,1h)8.02(br t,j=6.0hz,1h)。
[0423]
化合物13
[0424][0425]
相应地,化合物13以与化合物12相同的方式制备,从2-溴-6-甲基咪唑并[2,3-b][1,3]噻唑-5-甲酸cas[86933-042]和(4-{2-氮杂螺[3.3]庚-2-基}苯基)甲胺cas[1508720-12-4]开始,产量41mg,55%。
[0426]1h nmr(500mhz,dmso-d6)δppm 1.19(t,j=7.4hz,2h)1.70-1.86(m,2h)2.15(t,j=7.6hz,4h)2.42(d,j=1.3hz,3h)2.84(q,j=7.6hz,2h)3.72(s,4h)4.34(d,j=6.0hz,2h)6.36(d,j=8.5hz,2h)7.14(d,j=8.5hz,2h)7.88(d,j=1.3hz,1h)8.02(br t,j=6.0hz,1h)。
[0427]
化合物14的合成
[0428][0429]
中间体ai的制备
[0430]
将三乙胺(0.096ml,0.690mmol)、叔-丁基3-(氨甲基)-2-氧杂-9-氮杂螺[5.5]十一烷-9-甲酸酯(cas[1160246-99-0],100mg,0.352mmol)、hobt(46.6mg,0.345mmol)和edci
·
hcl(99.3mg,0.518mmol)依次添加至6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],77.5mg,0.345mmol)在二氯甲烷(2ml)中的溶液中。在60℃下搅拌16小时后,添加乙酸乙酯(20ml)。将混合物用水(2x20ml)和盐水(20ml)洗涤。将分离的有机层经硫酸钠干燥,过滤并将该滤液在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯1/1至0/1)进行纯化以给出中间体ai(160mg,产率:86%)。
[0431]
中间体aj的制备
[0432]
在0℃下,将盐酸盐(2ml,8mmol,2m在二噁烷中)添加至中间体ai(120mg,0.244mmol)在二氯甲烷(2ml)中的溶液中。在15℃下搅拌12小时后,将溶剂在真空下蒸发。将残余物溶于水(20ml)中,并且然后用饱和碳酸钠水溶液碱化至ph约为10。将溶液用二氯甲烷/甲醇(10/1,2x20ml)萃取。将合并的有机层用盐水(20ml)洗涤,经硫酸钠干燥,过滤,并且将滤液在真空下浓缩,以给出中间体aj(50mg,产率:56%)。
[0433]
化合物14的制备
[0434]
在n2气氛下,将中间体aj(30.0mg,0.0770mmol)、1-碘-4-(三氟甲氧基)苯(cas[103962-05-6],22.2mg,0.0770mmol)、pd(dba)2(4.60mg,8.00μmol)、x-phos(7.63mg,16.0μmol)和叔丁醇钠(29.6mg,0.308mmol)在二噁烷(4ml)中的溶液在110℃下在微波下辐射1小时。将该混合物进行过滤,并且将滤液在减压下浓缩。将残余物溶于乙酸乙酯中,用水、盐水洗涤,经na2so4干燥,过滤并减压浓缩至干燥。将残余物经硅胶柱层析法进行纯化(石油醚/乙酸乙酯10/1至0/1),以给出粗化合物。将其经gemini c18 150
×
25mm
×
10μl(洗脱液:
0.5%氨在水/乙腈中45/55至15/85)通过高效液相层析法进一步纯化。将这些所希望的级分进行收集并且进行冻干以给出化合物14(1.30mg,产率:3%)。
[0435]1h nmr(400mhz,cdcl3)δ=9.50(d,j=1.3hz,1h),7.54(d,j=9.5hz,1h),7.29(dd,j=2.1,9.4hz,1h),7.10(d,j=8.5hz,2h),6.89(d,j=9.3hz,2h),6.35(br.s.,1h),3.93-3.85(m,2h),3.51(br.s.,1h),3.30(m,1h),3.24(d,j=11.3hz,1h),3.21-3.07(m,4h),3.03(q,j=7.5hz,2h),1.94-1.85(m,2h),1.79(d,j=7.0hz,1h),1.66-1.62(m,1h),1.61-1.59(m,2h),1.50-1.47(m,2h),1.44(t,j=7.5hz,3h)
[0436]
化合物15的合成
[0437][0438]
中间体ak的制备
[0439]
在氮气气氛下,在10℃至15℃下,经一个小时将在二甲氧基乙烷(5ml)和丁醇(5ml)中的叔丁醇钠(481mg,5.01mmol)添加至7-boc-7-氮杂螺[3.5]壬-2-酮(cas[203661-69-2],600mg,2.51mmol)和tosmic(548mg,2.81mmol)在二甲氧基乙烷(5ml)中的溶液中。在20℃下搅拌混合物12小时后,将反应混合物倒入冰水中,并且然后用乙酸乙酯萃取。将萃取物用盐水洗涤,干燥并且蒸发。通过硅胶柱层析法(20%乙酸乙酯-己烷)纯化残余物以给出中间体ak(50.0mg,产率:8%)。
[0440]
中间体al的制备
[0441]
将中间体ak(50mg,0.200mmol)在nh3·
meoh(7m在甲醇中,10ml)中的溶液用拉尼镍(25mg)作为催化剂在15℃下氢化(h2,15psi)16小时。过滤掉催化剂,并且在真空下浓缩滤液以给出中间体al(50.9mg,产率:95%)。
[0442]
中间体am的制备
[0443]
将中间体al(44.9mg,0.200)、6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],50.9mg,0.200mmol)、hobt(27.0mg,0.200mmol)、edci(57.5mg,
0.300mmol)和三乙胺(0.056ml,0.400mmol)在dmf(2ml)中的溶液在60℃下搅拌16小时。添加乙酸乙酯(20ml)并且将混合物用盐水洗涤,干燥,过滤并且将滤液浓缩。将残余物经硅胶柱层析法(石油/乙酸乙酯1/1)进行纯化以给出中间体am(50.0mg,产率:51%)。
[0444]
中间体an的制备
[0445]
在0℃下,将盐酸盐(1.00ml,4.00mmol,4m在乙酸乙酯中)添加至中间体am(50.0mg,0.108mmol)在c中的溶液中。将混合物升温至20℃并搅拌16小时。用饱和碳酸钠中和混合物至ph约为10,并且用乙酸乙酯(10ml)稀释。将有机层用盐水(10ml)洗涤,经硫酸钠干燥,过滤并且在真空下浓缩。将残余物经硅胶通过薄层层析法(洗脱液:二氯甲烷/甲醇10/1)进行纯化以给出中间体an(35.0mg,产率:81%)。
[0446]
化合物15的制备
[0447]
在n2气氛下,将中间体an(15.0mg,0.0420mmol)、1-碘-4-(三氟甲氧基)苯(cas[103962-05-6],12.1mg,0.042mmol)、pd(dba)2(3.66mg,6.37μmol)、x-phos(3.81mg,8.00mmol)和叔丁醇钠(16.1mg,0.168mmol)在1,4-二噁烷(2ml)中的溶液在110℃下在微波下辐射60分钟。将该混合物进行过滤,并且然后将滤液在减压下浓缩。将残余物经硅胶柱层析法进行纯化(洗脱液:石油醚/乙酸乙酯1/1),以给出粗化合物。将其经geminic18 150
×
25mm
×
10μl(洗脱液:氨在水/乙腈中30/70至0/100)通过高效液相层析法进一步纯化。将这些所希望的级分进行收集并且进行冻干以给出化合物15(2.30mg,产率:10%)。
[0448]1h nmr(400mhz,cdcl3)δ=9.47(s,1h),7.54(d,j=9.5hz,1h),7.29(dd,j=2.0,9.5hz,1h),7.08(d,j=9.0hz,2h),6.89(d,j=9.0hz,2h),5.80(br.s.,1h),3.57(t,j=6.5hz,2h),3.17-3.09(m,2h),3.09-3.03(m,2h),3.00(q,j=7.5hz,2h),2.61(td,j=8.3,16.1hz,1h),2.11-1.99(m,2h),1.83-1.75(m,2h),1.71(d,j=5.5hz,2h),1.60-1.54(m,2h),1.45(t,j=7.7hz,3h)
[0449]
化合物16、化合物17、化合物18和化合物19的合成
[0450][0451]
中间体ao的制备
[0452]
在氮气气氛下,在0℃下,将dast(0.507ml,3.84mmol)逐滴添加至叔-丁基6-羟基-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[63711570],700mg,3.28mmol)在干燥的二氯甲烷(5ml)中的溶液中。将该混合物缓慢地升温至40℃并且搅拌过夜。将所得混合物用水和盐水洗涤。将有机层经硫酸镁干燥,过滤并且在真空下浓缩。将残余物经硅胶柱层析法进行纯化以给出中间体ao(200mg,产率:27%)。
[0453]
中间体ap的制备
[0454]
将中间体ao(200mg,0.929mmol)在甲酸(5ml)中的混合物在25℃下搅拌16小时。将混合物在真空下浓缩,以给出中间体ap(149mg,产率:100%)。
[0455]
化合物16的制备
[0456]
在n2下,将中间体ap(59.4mg,0.369mmol)、中间体m(195mg,0.443mmol)、pd(dba)2(21.2mg,0.037mmol)、x-phos(35.2mg,0.074mmol)和叔丁醇钠(177mg,1.85mmol)在1,4-二噁烷(8ml)中的溶液在110℃下在微波下辐射60分钟。添加二氯甲烷(50ml)并且将混合物用水(50ml)和盐水(50ml)洗涤。将有机层经硫酸钠干燥,过滤并将该滤液在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯1/0至0/1)进行纯化。将所希望的部分收集并且浓缩。经沃特斯xbridge c18 150
×
20mm
×
5μm(洗脱液:0.5%nh3水/甲醇35/65至5/95)通过高效液相层析法进一步纯化残余物。将这些所希望的级分进行收集并且进行冻干以给出化合物16(33.30mg,产率:21%)。
[0457]1h nmr(400mhz,cdcl3)δ=9.52(d,j=1.5hz,1h),7.53(d,j=9.5hz,1h),7.29(dd,j=2.1,9.4hz,1h),7.22(d,j=8.3 hz,2h),6.43(d,j=8.3 hz,2h),5.99(br.s.,
6.45(m,2h)5.84(br.s.,1h)5.05-4.9(m,1h)4.54(s,2h)3.88(s,2h)3.84(s,2h)2.82(q,j=7.70hz,2h)2.60-2.70(m,2h)2.37-2.52(m,5h)1.29-1.36(m,3h)。
[0472]
化合物19的制备
[0473][0474]
相应地,化合物19以与化合物11相同的方式制备,从中间体ar和2-乙基-5h,6h,7h,8h-咪唑并[1,2-a]吡啶-3-甲酸cas[1529528-99-1]开始,产量32.0mg,产率:21.5%。
[0475]
1h nmr(400mhz,cdcl3)δppm7.18(d,j=8.28hz,2h)6.41(d,j=8.53hz,2h)5.81(br.s.,1h)4.86-5.10(m,1h)4.48(d,j=5.52hz,2h)4.22(t,j=5.90hz,2h)3.88(s,2h)3.84(s,2h)2.85(t,j=6.40hz,2h)2.60-2.70(m,4h)2.37-2.51(m,2h)1.83-1.99(m,4h)1.22(t,j=7.65hz,3h)
[0476]
化合物20和化合物21的合成
[0477][0478]
中间体as的制备
[0479]
将tfa(1.6ml,21mmol)添加至叔-丁基6-氧杂-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1181816-12-5],0.3g,1.4mmol)在二氯甲烷(9.8ml)中的溶液中并且将混合物在室温下搅拌18小时。将反应混合物在真空下蒸发,并与甲苯共蒸发两次以给出呈无色油状物(100%)的320mg中间体as。
[0480]
中间体at的制备
[0481]
用功率输出范围从0至400w的单模式微波(biotage initiator 60),将中间体as
(0.34g,1.5mmol)、4-氟苯甲腈(cas[1194-02-1],0.37g,3.0mmol)和k2co3(0.62g,4.5mmol)在dmso(5.4ml)中的溶液在120℃下加热30分钟。添加盐水和etoac。将有机层萃取,经mgso4干燥,过滤并且蒸发。通过制备型lc(因特奇美拉,12g,30μm,庚烷/etoac 90/10)进行残余物的纯化。收集纯的级分并且蒸发,以给出呈白色固体(19%)的60mg中间体at。
[0482]
中间体au的制备
[0483]
在0℃下,将中间体at(60mg,0.28mmol)在干燥的thf(1.1ml)中的溶液逐滴添加至lialh4(64mg,1.7mmol)在干燥的thf(1.2ml)中的混合物中。使混合物缓慢回到室温下并搅拌过夜。十分缓慢地添加水(0.24ml),然后十分缓慢添加二氯甲烷(30ml)并且搅拌20分钟。添加mgso4,用硅藻土垫过滤不溶物,并且将滤液蒸发至干燥以给出57mg呈白色固体(92%)的中间体au。
[0484]
化合物20的制备
[0485]
将6-乙基-2-甲基咪唑并[2,1-b]噻唑-5-甲酸(cas[1131613-58-5],46mg,0.22mmol)、中间体au(57mg,0.26mmol)、edci
·
hcl(34mg,0.22mmol)、hobt(29mg,0.22mmol)和dipea(0.038ml,0.22mmol)在二氯甲烷(1.5ml)和thf(1.5ml)中的溶液在室温下搅拌18小时。将混合物用硅胶扩展并且在真空中蒸发。通过制备型lc(常规sioh30μm,12g,干法填充,流动相梯度:dcm/meoh,从99/1至96/4)纯化残余物以在蒸发、在et2o中研磨以及第二次蒸发后给出53mg呈米色固体的化合物20(59%)。
[0486]1h nmr(400mhz,dmso-d6)δppm 1.19(t,j=7.6hz,3h)1.89-2.02(m,2h)2.38-2.45(m,2h)2.41(s,3h)2.83(q,j=7.6hz,2h)3.67(s,2h)3.72(s,2h)3.90-4.08(m,1h)4.34(d,j=5.6hz,2h)5.01(d,j=6.6hz,1h)6.34(d,j=8.6hz,2h)7.13(d,j=8.1hz,2h)7.87(d,j=1.0hz,1h)7.99(t,j=6.1hz,1h)。
[0487]
化合物21的制备
[0488]
在氮气下,将在二氯甲烷(0.20ml,94μmol)中的dmp(15%)添加至化合物20(35mg,85μmol)在二氯甲烷(2.7ml)中的溶液中并且将混合物在室温下搅拌72小时。将混合物用硅胶扩展并且在真空中蒸发。通过制备型lc(常规sioh 30μm,12g,干法填充,流动相梯度:dcm/meoh,从99/1至97/3)纯化残余物以在蒸发、在et2o中研磨和蒸发后给出18mg的米色固体。通过反相(固定相:x-bridge-c18 5μm30*150mm,流动相:梯度从75%nh4hco3水溶液(0.5%)、25%mecn到35%nh4hco3水溶液(0.5%)、65%mecn)纯化固体以给出5mg呈米色固体(14%)的化合物21。
[0489]1h nmr(400mhz,dmso-d6)δppm 1.19(t,j=7.6hz,3h)2.41(s,3h)2.84(q,j=7.6hz,2h)3.32(s,4h)3.95(s,4h)4.35(d,j=5.6hz,2h)6.43(d,j=8.1hz,2h)7.17(d,j=8.6hz,2h)7.88(s,1h)8.02(t,j=5.6hz,1h)。
[0490]
化合物23和化合物22的合成
[0491][0492]
中间体av的制备
[0493]
相应地,中间体av以与中间体s相同的方式制备,从中间体r和苯基硼酸cas[98-80-6]开始,产量0.3g,62%。
[0494]
中间体aw的制备
[0495]
相应地,中间体aw以与中间体t相同的方式制备,从中间体av开始,产量0.27g,99%。
[0496]
化合物22和化合物23的制备
[0497]
相应地,化合物22以与化合物7相同的方式制备,从中间体aw和中间体m开始,产出0.031g,16%的化合物22,和0.0071g,13%作为副产物的化合物23。
[0498]
化合物221h nmr(400mhz,氯仿-d)δ=9.53(d,j=2.0hz,1h),7.57-7.49(m,1h),7.28(d,j=2.3hz,3h),7.24(s,1h),7.23-7.17(m,4h),6.47(d,j=8.3hz,2h),5.98(br.s.,1h),4.58(d,j=5.5hz,2h),4.02(s,2h),3.81(s,2h),3.48(quin,j=8.9hz,1h),2.94(q,j=7.5hz,2h),2.69-2.60(m,2h),2.41-2.32(m,2h),1.38(t,j=7.7hz,3h)。
[0499]
化合物231h nmr(400mhz,氯仿-d)δppm9.40(d,j=7.28hz,1h)7.60(d,j=9.03hz,1h)7.34-7.31(m,3h)7.29-7.20(m,5h)6.88-6.95(m,1h)6.47(d,j=8.28hz,2h)5.97(br.s.,1h)4.59(d,j=5.27hz,2h)4.02(s,2h)3.81(s,2h)3.38-3.54(m,1h)2.96(q,j=7.61hz,2h)2.59-2.74(m,2h)2.31-2.42(m,2h)1.39(t,j=7.53hz,3h)
[0500]
化合物24的合成
[0501][0502]
中间体ax的制备
[0503]
将6-甲基咪唑并[2,1-b][1,3]噻唑-5-甲酸(cas[77628-51-4],200mg,1.10mmol)、4-碘苯甲胺(cas[39959-59-6],256mg,1.10mmol)、hatu(544mg,1.43mmol)、和二异丙基乙胺(425mg,3.29mmol)在二氯甲烷(5ml)中的混合物在25℃下搅拌2小时。将混合物用二氯甲烷(100ml)稀释。将溶液用水(50ml)、盐水(50ml)洗涤,经硫酸钠干燥,过滤并在真空下浓缩。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯0/1)进行纯化以给出中间体ax(220mg,产率:47.3%)。
[0504]
化合物24的制备
[0505]
相应地,从中间体ax和中间体t开始以与化合物7相同的方式制备化合物24,产量0.029g,17%。
[0506]1h nmr(400mhz,氯仿-d)δppm 8.29(d,j=4.5hz,1h),7.24-7.18(m,4h),7.18-7.13(m,2h),6.88(d,j=4.5hz,1h),6.46(d,j=8.5hz,2h),5.85(br.s.,1h),4.56(d,j=5.5hz,2h),4.02(s,2h),3.80(s,2h),3.47(quin,j=8.9hz,1h),2.70-2.61(m,2h),2.56(s,3h),2.38-2.29(m,2h)
[0507]
化合物25和化合物26的合成
[0508][0509]
中间体ay的制备
[0510]
将2-氯-6-喹啉甲腈(cas[78060-54-5],14.7mg,0.078mmol)、中间体t(20.0mg,0.078mmol)和碳酸钾(21.6mg,0.156mmol)在乙腈(5ml)中的混合物回流16小时。在真空下将溶剂蒸发。将残余物经硅胶柱层析法(洗脱液:石油醚/乙酸乙酯1/1)进行纯化以给出中间体ay(20.0mg,产率:62.8%)。
[0511]
中间体az的制备
[0512]
将中间体ay(20.0mg,0.049mmol)在nh3·
meoh(20ml,7m nh3在meoh)中的溶液在15℃下(15psi)用拉尼镍(3mg)作为催化剂氢化16小时。过滤掉催化剂,并且在真空下浓缩滤液以给出中间体az(20.0mg,产率:91.84%)。
[0513]
化合物26的制备
[0514]
在25℃下,将6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],9.79mg,0.044mmol)、hatu(21.7mg,0.057mmol)、diea(14.8mg,0.114mmol)在ch2cl2(10ml)中的溶液搅拌30分钟。将中间体az(20mg,0.048mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:0.05%氨在水/甲醇中35/65至5/95)。收集所需级分并浓缩,以给出化合物26(4.30mg,15.91%)。
[0515]
1h nmr(400mhz,氯仿-d)δppm 9.56(s,1h)7.84(d,j=8.80hz,1h)7.74(d,j=8.56hz,1h)7.59(s,1h)7.55(d,j=9.29hz,2h)7.31(d,j=9.78hz,1h)7.22(d,j=8.40hz,2h)7.16(d,j=8.40hz,2h)6.59(d,j=9.05hz,1h)6.13(br.s.,1h)4.79(d,j=5.62hz,2h)4.33(s,2h)4.11(s,2h)3.50(t,j=8.68hz,1h)2.97(q,j=7.42hz,2h)2.65-2.76(m,2h)2.33-2.44(m,2h)1.38(t,j=7.58hz,3h)
[0516]
化合物25的制备
[0517]
相应地,从中间体l和中间体az开始以与化合物26相同的方式制备化合物25,产量0.037g,25%。
[0518]
1h nmr(400mhz,氯仿-d)δppm 9.83(d,j=2.51hz,1h)8.55(d,j=2.51hz,1h)7.83(d,j=8.78hz,1h)7.75(d,j=8.53hz,1h)7.55(d,j=9.20hz,1h)7.53(d,j=6.80hz,1h)7.22(d,j=8.80hz,2h)7.15(d,j=8.40hz,2h)6.58(d,j=8.78hz,1h)6.26(t,j=5.27hz,1h)4.77(d,j=5.60hz,2h)4.33(s,2h)4.11(s,2h)3.50(quin,j=8.85hz,1h)3.01(q,j=7.53hz,2h)2.65-2.74(m,2h)2.33-2.43(m,2h)1.41(t,j=7.53hz,3h)
[0519]
还根据在此描述的这些程序来制备以下化合物:
[0520]
[0521]
[0522]
[0523]
[0524]
[0525][0526]
化合物56的合成
[0527][0528]
向5-甲氧基-2-甲基吡唑并[1,5-a]吡啶-3-甲酸(cas[1352395-28-8],0.055g mg,0.26mmol)在dmf(5ml)中的溶液中添加中间体i(0.08g,0.22mmol)、hatu(0.1g,0.26mmol)和二异丙基乙胺(0.085g,0.66mmol)。将混合物在室温下搅拌过夜。将溶剂在真空中除去至干燥。通过高效液相层析法(沃特斯xbridge prep obd c18 150x30x5μ,25ml/min,梯度水(含有0.05%nh3.h2o)/乙腈从85/15至55/45)纯化残余物。收集所需的级分并在真空中蒸发以除去乙腈。将残余物冻干以给出0.027g,21%的化合物56。
[0529]1h nmr(400mhz,cdcl3)δ=8.19(d,j=7.5hz,1h),7.57(d,j=2.2hz,1h),7.34(d,j=7.5hz,2h),7.21(d,j=7.9hz,2h),7.07(d,j=8.4hz,2h),6.54(dd,j=2.4,7.3hz,1h),6.40(d,j=8.8hz,2h),5.96(br.s.,1h),4.67(d,j=5.3hz,2h),4.01(s,2h),3.91(s,3h),3.80(s,2h),3.51-3.44(m,1h),2.70-2.56(m,5h),2.41-2.30(m,2h)。
[0530]
化合物57的合成
[0531]
[0532]
相应地,化合物57以与化合物56相同的方式制备,从5-甲氧基-2-甲基吡唑并[1,5-a]吡啶-3-甲酸cas[1352395-28-8]和中间体q开始,产量0.027g,21%。
[0533]
1h nmr(400mhz,cdcl3)δ=8.18(d,j=7.5hz,1h),7.57(d,j=2.6hz,1h),7.25(br.s.,2h),7.09(d,j=8.8hz,2h),6.53(dd,j=2.6,7.5hz,1h),6.49(d,j=8.4hz,2h),6.43(d,j=8.8hz,2h),5.86(br.s.,1h),4.59(d,j=5.3hz,2h),4.04(s,8h),3.91(s,3h),2.58(s,3h)
[0534]
化合物58的合成
[0535][0536]
中间体ba的制备
[0537]
将2-氨基-5-氯吡嗪(cas[33332-29-5],6g,46.31mmol)和中间体j(14.52g,69.47mmol)在etoh(10ml)中的混合物在100℃下搅拌12小时。在真空中将溶剂除去。将该残余物通过柱层析法(石油醚/乙酸乙酯=5/1)进行纯化。收集产物级分并蒸发溶剂以获得中间体ba,0.81g,70%。
[0538]
中间体bb的制备
[0539]
向中间体ba(0.8g,3.34mmol)在meoh(30ml)和水(6ml)中的溶液中添加氢氧化锂一水合物(0.7g,16.69mmol)。将该混合物在室温下搅拌10小时。在真空中将溶剂除去。将混合物用水性hcl 2n(5ml)酸化至ph=3-4。过滤出所得的白色沉淀物,并且用水(20ml)洗涤以给出中间体bb,0.65g,86%。
[0540]
化合物58的制备
[0541]
相应地,从中间体bb和中间体q开始以与化合物56相同的方式制备化合物58,产量0.05g,29%。
[0542]
1h nmr(400mhz,cdcl3)δ=9.41(s,1h),8.90(s,1h),7.24(d,j=7.9hz,2h),7.08(d,j=8.4hz,2h),6.49(d,j=8.4hz,2h),6.42(d,j=8.8hz,2h),6.10(br.s.,1h),4.60(d,j=5.3hz,2h),4.04(d,j=3.5hz,8h),3.00(q,j=7.5hz,2h),1.42(t,j=7.5hz,3h)
[0543]
化合物59的合成
[0544][0545]
中间体bc的制备
[0546]
将硼氢化钠(2.13g,56.41mmol)添加至7-boc-7-氮杂螺[3.5]壬-2-酮(cas[203661-69-2],2.5g,10.45mmol)在meoh(30ml)中的溶液中。将该混合物在25℃下搅拌16小、时。将混合物在真空下浓缩。将残余物用乙酸乙酯(50ml)稀释,用水(2x50ml)和盐水(50ml)洗涤。将分离的有机层经无水硫酸钠干燥并在真空下浓缩以给出中间体bc,2.5g,99%。
[0547]
中间体bd的制备
[0548]
在0℃下,将hcl 4m在etoac(5.18ml,20.72mmol)中的溶液添加至中间体bc(2.5g,10.36mmol)在ch2cl2(100ml)的溶液中。将该溶液在室温下搅拌过夜。将溶剂在真空下浓缩,给出呈盐酸盐的中间体bd,1.84g,100%。
[0549]
中间体be的制备
[0550]
向1-碘-4-(三氟甲氧基)苯(cas[103962-05-6],4.48g,15.54mmol)在dmso(50ml)中的溶液中添加中间体bd(1.84g,10.36mmol)、碳酸铯(8.44g,25.9mmol)、l-脯氨酸(0.48g,4.14mmol)和碘化亚铜(0.39g,2.07mmol)。在氩气氛下将混合物在90℃下加热18小时。将混合物用水(100ml)稀释并用乙酸乙酯萃取(50mlx3)。将有机层用盐水(50ml)洗涤,经na2so4干燥,过滤,并在真空中浓缩。将该残余物通过柱层析法(石油醚/乙酸乙酯=4/1)
纯化以给出中间体be,1.5g,48%。
[0551]
中间体bf的制备
[0552]
将甲磺酰氯(0.77ml,9.96mmol)添加至中间体be(1.5g,4.98mmol)和三乙胺(2.78ml,19.91mmol)在ch2cl2(20ml)中的溶液中。该反应溶液在室温下搅拌过夜。将混合物用水(100ml)洗涤并且在真空下浓缩。将该残余物通过硅胶柱层析法(石油醚:乙酸乙酯4/1)纯化。收集纯的级分并蒸发以给出中间体bf,1.6g,85%。
[0553]
中间体bg的制备
[0554]
将中间体bf(1.6g,4.22mmol)、氰化钠(0.83g,16.87mmol)和四丁基溴化铵(0.82g,2.53mmol)在dmf(30ml)中的混合物在120℃下搅拌10小时。将混合物用水(200ml)稀释并用乙酸乙酯萃取(200mlx3)。将有机层用盐水(200ml)洗涤,经na2so4干燥,过滤,并在真空中浓缩。将该残余物通过硅胶柱层析法(石油醚:乙酸乙酯4/1)纯化。收集产物级分并蒸发溶剂,给出中间体bg,1.3g,99%。
[0555]
中间体bh的制备
[0556]
将中间体bg(1.3g,4.19mmol)在nh3·
meoh(7m在甲醇中,20ml)中的混合物用拉尼镍(1g)作为催化剂在25℃下氢化(15psi)16小时。在吸收了h2之后,将催化剂过滤出,并且将滤液进行浓缩以给出中间体bh,1.3g,99%。
[0557]
化合物59的制备
[0558]
相应地,化合物59以与化合物56相同的方式制备,从5-甲氧基-2-甲基吡唑并[1,5-a]吡啶-3-甲酸cas[1352395-28-8]和中间体bh开始,产量0.048g,36%。
[0559]
1h nmr(400mhz,cdcl3)δ=8.18(d,j=7.5hz,1h),7.54(d,j=2.6hz,1h),7.08(d,j=8.8hz,2h),6.88(d,j=8.8hz,2h),6.53(dd,j=2.6,7.5hz,1h),5.66(br.s.,1h),3.90(s,3h),3.53(t,j=6.4hz,2h),3.16-3.09(m,2h),3.08-3.02(m,2h),2.63-2.60(m,3h),2.04(t,j=10.4hz,2h),1.81-1.75(m,2h),1.72-1.66(m,2h),1.62(br.s.,2h)
[0560]
化合物60的合成
[0561][0562]
相应地,从中间体l和中间体bh开始以与化合物59相同的方式制备化合物60,产量0.075g,45%。
[0563]1h nmr(400mhz,cdcl3)δ=9.79(d,j=2.2hz,1h),8.56(d,j=2.2hz,1h),7.09(d,j=8.8hz,2h),6.89(d,j=8.8hz,2h),5.86(br.s.,1h),3.57(t,j=6.4hz,2h),3.17-3.11(m,2h),3.10-3.00(m,4h),2.61(td,j=8.0,16.2hz,1h),2.11-2.01(m,2h),1.83-1.77(m,2h),1.75-1.68(m,2h),1.64(m,2h),1.49(t,j=7.5hz,3h).
[0564]
化合物61的合成
[0565][0566]
相应地,化合物61以与化合物59相同的方式从2-乙基-5h,6h,7h,8h-咪唑并[1,2-a]吡啶-3-甲酸cas[1529528-99-1]和中间体bh开始制备,产量0.082g,65%。
[0567]1h nmr(400mhz,cdcl3)δ=7.09(d,j=8.5hz,2h),6.96-6.85(m,2h),5.64(br.s.,1h),4.20(t,j=5.9hz,2h),3.47(dd,j=5.8,7.3hz,2h),3.16-3.09(m,2h),3.08-3.02(m,2h),2.86(t,j=6.3hz,2h),2.73(q,j=7.6hz,2h),2.55(td,j=7.9,16.0hz,1h),2.05-1.98(m,2h),1.97-1.85(m,4h),1.82-1.74(m,2h),1.71-1.67(m,2h),1.61-1.52(m,2h),1.30(t,j=7.7hz,3h)。
[0568]
化合物62的合成
[0569][0570]
中间体bi的制备
[0571]
在n2流下,在-70℃下,将lihmds(19.27ml,19.27mmol)添加至在thf(180ml)中的二乙基氰基甲基膦酸酯(3.41g,19.27mmol)的混合物中。将混合物搅拌10分钟。在-78℃下,将1-[4-(三氟甲氧基)苯基]-4-哌啶酮(cas[681508-68-9],4.5g,17.36mmol)添加至混合物。将该混合物在-78℃搅拌1小时。将混合物用nh4cl溶液淬灭,用乙酸乙酯(300ml)萃取,用盐水(200ml)洗涤,经mgso4干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(乙酸乙酯/石油醚为从0至1/3)进行纯化。将所希望的级分收集并浓缩以给出7.8g,72%的中间体bi。
[0572]
中间体bj的制备
[0573]
将碘化三甲基氧化锍(5.83g,26.5mmol)缓慢地添加至叔丁醇钾(2.97g,26.5mmol)在dmso(50ml)中的溶液中。将该混合物在室温下搅拌1.5小时。将中间体bi
(6.8g,24.09mmol)在dmso(50ml)中的溶液添加至混合物中。将混合物在45℃下搅拌24小时。将饱和nh4cl溶液添加至混合物中并且搅拌0.5小时。将该混合物用乙酸乙酯(100ml)萃取。将有机层用盐水(70ml)洗涤,经mgso4干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(乙酸乙酯/石油醚为从0至1/3)进行纯化。将所希望的级分收集并浓缩以给出4.5g,63%的中间体bj。
[0574]
中间体bk的制备
[0575]
因此,中间体bk以与中间体bh相同的方式制备,从中间体bj开始,给出0.18g,
[0576]
化合物62的制备
[0577]
相应地,化合物62以与化合物56相同的方式制备,从5-甲氧基-2-甲基吡唑并[1,5-a]吡啶-3-甲酸cas[1352395-28-8]和中间体bk开始,产量0.04g,9%。
[0578]
1h nmr(400mhz,cdcl3)δ=8.18(d,j=7.1hz,1h),7.53(d,j=2.2hz,1h),7.10(d,j=8.8hz,2h),6.91(d,j=9.3hz,2h),6.52(dd,j=2.4,7.3hz,1h),5.73(br.s.,1h),3.89(s,3h),3.60-3.48(m,2h),3.34(t,j=13.0hz,2h),3.17-3.09(m,2h),2.63(s,3h),1.93-1.84(m,1h),1.80-1.73(m,1h),1.66-1.58(m,1h),1.42-1.34(m,1h),1.10-1.00(m,1h),0.67(dd,j=4.6,8.2hz,1h),0.36(t,j=4.9hz,1h)
[0579]
化合物63的合成
[0580][0581]
相应地,从中间体l和中间体bk开始以与化合物62相同的方式制备化合物63,产量0.019g,16%。
[0582]
1h nmr(400mhz,cdcl3)δppm 9.78(d,j=2.76hz,1h)8.56(d,j=2.76hz,1h)7.11(d,j=8.28hz,2h)6.86-6.96(m,2h)5.92(br.s.,1h)3.57(dd,j=7.65,5.40hz,2h)3.30-3.43(m,2h)3.09-3.17(m,2h)3.06(q,j=7.53hz,2h)1.90(ddd,j=12.92,9.16,3.26hz,1h)1.74-1.83(m,1h)1.63(br.s.,1h)1.47(t,j=7.65hz,3h)1.30-1.41(m,1h)0.99-1.10(m,1h)0.71(dd,j=8.28,4.77hz,1h)0.38(t,j=5.02hz,1h)
[0583]
化合物64的合成
[0584][0585]
相应地,化合物64以与化合物62相同的方式制备,从6-乙基-2-甲基咪唑并[2,1-b]噻唑-5-甲酸cas[1131613-58-5]和中间体bk开始,产量0.048g,32%。
[0586]
1h nmr(400mhz,cdcl3)δppm 7.95(d,j=1.51hz,1h)7.10(d,j=8.53hz,2h)6.91
(d,j=8.53hz 2h)5.72(br.s.,lh)3.45-3.58(m,2h)3.34(t,j=13.05hz,2h)3.07-3.18(m,2h)2.89(q,j=7.53hz,2h)2.43(d,j=1.51hz,3h)1.82-1.93(m,1h)1.69-1.80(m,1h)1.63(br.s.,1h)1.38(t,j=7.65hz,3h)1.30(t,j=7.65hz,1h)0.97-1.07(m,1h)0.68(dd,j=8.91,4.39hz,1h)0.35(t,j=4.89hz,1h)
[0587]
化合物65的合成
[0588][0589]
在25℃下,将2-乙基-5h,6h,7h,8h-咪唑并[1,2-a]吡啶-3-甲酸(cas[1529528-99-1],0.18g,0.41mmol)、hatu(0.204g,0.54mmol)、二异丙基乙胺(0.139g,1.08mmol)在dmf(5ml)中的溶液搅拌30分钟。将中间体i(0.15g,0.41mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。通过沃特斯xbridge prep obd高效液相层析法(洗脱液:0.05%氨水/乙腈25/75至5/95)纯化粗产物。收集所需级分并冻干,以给出化合物65,0.035g,29%。
[0590]
1h nmr(400mhz,cdcl3)δppm 7.27-7.31(m,2h)7.19(d,j=8.03hz,2h)7.06(d,j=8.28hz,2h)6.37-6.42(m,2h)5.92(br.s.,1h)4.58(d,j=5.77hz,2h)4.23(t,j=5.77hz,2h)4.01(s,2h)3.79(s,2h)3.47(quin,j=8.72hz,1h)2.86(t,j=6.40hz,2h)2.71(q,j=7.70hz,2h)2.61-2.68(m,2h)2.31-2.39(m,2h)1.83-2.00(m,4h)1.25(t,j=7.65hz,3h)
[0591]
化合物66的合成
[0592][0593]
将中间体l(0.011g,0.049mmol)、中间体ad(0.02g,0.049mmol)、hatu(0.024g,0.063mmol)、和二异丙基乙胺(0.032g,0.245mmol)在二氯甲烷(1ml)中的混合物在室温下搅拌2小时。将混合物在真空下浓缩。添加乙酸乙酯(20ml)并且将混合物用水(2x20ml)和盐
水(20ml)洗涤。将分离的有机层经硫酸镁干燥,过滤并且在真空下浓缩。经菲罗门gemini c18 250
×
21.2mm
×
5μm,通过高效液相层析法(洗脱液:水(0.05%氢氧化铵v/v)/甲醇25/75至5/95)纯化残余物。收集所需级分并冻干,以给出化合物66,0.011g,37%。
[0594]
化合物67的合成
[0595][0596]
在25℃下,将6-乙基-2-甲基咪唑并[2,1-b]噻唑-5-甲酸(cas[1131613-58-5],0.038g,0.18mmol)、hatu(0.082g,0.22mmol)、和二异丙基乙胺(0.056g,0.43mmol)在dmf(20ml)中的混合物搅拌30分钟。将中间体i(0.06g,0.17mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。经菲罗门gemini,通过高效液相层析法(水(0.05%hcl)/acn 60/40至30/70)纯化粗产物。收集所需级分并冻干,以给出化合物67,0.036g,33%。
[0597]1h nmr(400mhz,氯仿-d)δ=7.98(s,1h),7.28(d,j=7.5hz,2h),7.18(d,j=7.9hz,2h),7.04(d,j==8.8hz,2h),6.38(d,j=8.8hz,2h),5.96(br.s.,1h),4.62(d,j=5.7hz,2h),3.99(s,2h),3.77(s,2h),3.45(quin,j=8.8hz,1h),2.84(q,j=7.5hz,2h),2.67-2.59(m,2h),2.47-2.38(m,3h),2.36-2.29(m,2h),1.40-1.29(m,3h)
[0598]
化合物68的合成
[0599][0600]
将二异丙基乙胺(0.512ml;2.98mmol)和hatu(0.588g;1.55mmol)依次添加至6-乙基-2-甲基咪唑并[2,1-b]噻唑-5-甲酸(cas[1131613-58-5],0.25g,1.19mmol)在dmf(30ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体q(0.432g,1.19mmol),并将混合物在室温下搅拌4小时。将反应混合物在真空中蒸发至干燥,用etoac稀释并用盐水洗涤(两次)。将有机层经mgso4干燥,过滤并且蒸发以给出1.1g的棕色油状物。通过制备型lc(常规sioh 30μm,40g因特奇美拉,干法填充流动相梯度:从ch2cl2/meoh 100:0至95:5)纯化粗产物,以得到0.203g灰白色泡沫,将其在et2o中研磨,过滤并在高真空下干燥以给出0.151g呈灰白色固体的化合物68(23%)。
[0601]1h nmr(400mhz,dmso-d6)δppm8.02(t,j=5.8hz,1h),7.87(d,j=1.5hz,1h),7.16
(dd,j=8.6,3.5hz,4h),6.49(d,j=8.0hz,2h),6.42(d,j=8.6hz,2h),4.35(d,j=6.1hz,2h),3.94(s,4h)4.00(s,4h),2.84(q,j=7.4hz,2h),2.41(d,j=1.5hz,3h),1.19(t,j=7.6hz,3h)。
[0602]
化合物69、化合物70和化合物71的合成
[0603][0604]
中间体bl的制备
[0605]
将2-氨基吡嗪(cas[5049-61-6],12g,126.18mmol)和中间体j(39.6g,189.27mmol)在etoh(10ml)中的混合物在100℃下搅拌12小时。在真空中将溶剂除去。将该粗产物通过柱层析法(石油醚/乙酸乙酯=5/1至1/1)进行纯化。收集产物级分并蒸发溶剂以给出中间体bl,2g,8%。
[0606]
中间体bm的制备
[0607]
在n2下,向中间体bl(5g,24.36mmol)在meoh(20ml)的溶液中添加二氧化普拉廷白铜(500mg),随后添加一滴浓盐酸。将悬浮液在真空下脱气并且用h2吹扫若干次。将该混合物在25℃在h2(15psi)下搅拌10小时。将悬浮液通过垫过滤,并将该垫用甲醇(50ml)洗涤。将合并的滤液浓缩至干燥以给出中间体bm,5g,98%。
[0608]
中间体bn的制备
[0609]
在0℃下,向中间体bm(5g,23.89mmol)在meoh(75ml)的溶液中添加甲醛水溶液(9.7g,119.47mmol,37%),随后添加硼氰基氢化钠(7.5g,119.47mmol)和一滴乙酸(0.2ml)。然后,将混合物在室温下搅拌过夜。逐滴添加10%nh4cl溶液(25ml)。将混合物用乙酸乙酯萃取,将合并的有机层用盐水洗涤,经na2so4干燥,过滤并且将溶剂在真空下蒸发。通过硅胶柱层析法(二氯甲烷/甲醇=15∶1至10∶1)纯化残余物以给出中间体bn,1.3g,24%。
[0610]
中间体bo的制备
[0611]
像中间体bn(0.55g,2.46mmol)在meoh(25ml)和水(5ml)中的溶液中添加氢氧化锂一水合物(0.52g,12.32mmol)。将该混合物在室温下搅拌10小时。将溶剂在真空中除去至干燥。通过高效液相层析法(durashell 150
×
25mm
×
5μm,25ml/min,水(含有0.05%hcl)/乙腈从100/0至70/30)纯化残余物。收集所需的级分并在真空中蒸发以除去乙腈。将残余物冻干以给出0.4g,78%的中间体bo。
[0612]
化合物69的制备
[0613][0614]
在25℃下,将中间体bo(0.04g,0.19mmol)、hatu(0.095g,0.25mmol)、二异丙基乙胺(0.064g,0.5mmol)在dmf(5ml)中的溶液搅拌30分钟。将中间体i(0.069g,0.19mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。通过沃特斯xbridge prep obd高效液相层析(洗脱液:0.05%氨水/乙腈从50/50至20/80)纯化粗产物。收集所需级分并冻干,以给出化合物69,0.053g,50%。
[0615]
1h nmr(400mhz,cdcl3)δppm7.27-7.31(m,2h)7.17-7.22(m,2h)7.06(d,j=8.28hz,2h)6.37-6.42(m,2h)5.94(br.s.,1h)4.58(d,j=5.52hz,2h)4.32(t,j=5.65hz,2h)4.01(s,2h)3.79(s,2h)3.65(s,2h)3.39-3.53(m,1h)2.80(t,j=5.65hz,2h)2.72(q,j=7.70hz,2h)2.61-2.68(m,2h)2.47(s,3h)2.29-2.40(m,2h)1.26(t,j=7.53hz,3h)
[0616]
化合物70的制备
[0617][0618]
相应地,从中间体bo和中间体q开始以与化合物69相同的方式制备化合物70,给出0.06g,50%。
[0619]1h nmr(400mhz,cdcl3)δ=7.21(d,j=8.3hz,2h),7.09(d,j=8.3hz,2h),6.45(dd,j=8.5,17.8hz,4h),5.85(br.s.,1h),4.50(d,j=5.5hz,2h),4.32(t,j=5.4hz,2h),4.04(s,8h),3.65(s,2h),2.80(t,j=5.6hz,2h),2.70(q,j=7.4hz,2h),2.48(s,3h),1.24(t,j=7.5hz,3h)。
[0620]
化合物71的制备
[0621][0622]
相应地,从中间体bo和中间体v开始以与化合物69相同的方式制备化合物71,给出0.035g,38%。
[0623]
1h nmr(400mhz,cdcl3)δppm 7.10-7.23(m,6h)6.45(d,j=7.94hz,2h)5.83(br.s.,1h)4.48(d,j=5.29hz,2h)4.32(t,j=5.51hz,2h)4.01(s,2h)3.80(s,2h)3.64(s,2h)3.43-3.50(m,1h)2.79(t,j=5.51hz,2h)2.67(dt,j=15.33,7.99hz,4h)2.47(s,3h)2.28-2.39(m,2h)1.23(t,j=7.50hz,3h)
[0624]
化合物72、化合物73和化合物74的合成
[0625][0626]
中间体bp的制备
[0627]
在0℃下,在n2流中,将在甲苯(10ml)中的diad(1.40g,6.92mmol)添加至叔-丁基6-羟基-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1147557-97-8],1.2g,5.63mmol)、4-(三氟甲基)苯酚(cas[402-45-9],1.10g,6.75mmol)、和三苯基膦(2.31g,8.80mmol)在甲苯(40ml)中的溶液中。将该混合物在室温下搅拌过夜。将混合物浓缩。将该粗产物经硅胶柱层析法(石油醚/乙酸乙酯从1/0至3/1)进行纯化。将所希望的级分收集并浓缩以给出2g,99%的中间体bp。
[0628]
中间体bq的制备
[0629]
将中间体bp(2g,5.60mmol)在甲酸(10ml)中的混合物搅拌12小时。将混合物浓缩以给出中间体bq,1.4g,97%。
[0630]
中间体br的制备
[0631]
在n2流中,将中间体bq(1.4g,5.44mmol)、4-碘苯基氰(cas[3058-397],0.99g,5.44mmol)、binap(0.203g,0.33mmol)、pd2(dba)3(0.1g,0.11mmol)、叔丁醇钠(1.57g,16.33mmol)和三乙胺(0.38ml)在甲苯(50ml)中的溶液在110℃下搅拌过夜。将混合物浓缩。将残余物溶解于ch2cl2(100ml)和水(100ml)中。将有机层用盐水(100ml)洗涤,经mgso4干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(乙酸乙酯/石油醚从0至1/5)进行纯化。将所希望的级分收集并浓缩以给出1.8g,92%的中间体br。
[0632]
中间体bs的制备
[0633]
在25℃(15psi)下,将中间体br(0.2g,0.56mmol)于氨7n(在甲醇中)(20ml)的混合物用拉尼镍(20mg)作为催化剂氢化16小时。在吸收了h2之后,将催化剂过滤出,并且将滤液进行浓缩以给出中间体bs,0.2g,99%。
[0634]
化合物73的制备
[0635][0636]
在25℃下,将中间体l(0.112g,0.25mmol)、hatu(0.122g,0.32mmol)、二异丙基乙胺(0.083g,0.65mmol)在dmf(10ml)中的溶液搅拌30分钟。将中间体bs(0.09g,0.25mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。经菲罗门gemini,通过高效液相层析法(洗脱液:0.05%氨水/乙腈35/65至5/95)纯化粗产物。收集所需级分并冻干,以给出化合物73,0.016g,11%。
[0637]
1h nmr(400mhz,cdcl3)δppm 9.83(d,j=2.65hz,1h)8.47-8.60(m,1h)7.53(d,j=8.38hz,2h)7.22(d,j=7.94hz,2h)6.86(d,j=8.38hz,2h)6.45(d,j==8.38hz,2h)6.05(br.s.,1h)4.63-4.71(m,1h)4.58(d,j==5.29hz,2h)3.95(s,2h)3.90(s,2h)2.98(q,j=7.50hz,2h)2.76-2.84(m,2h)2.39-2.47(m,2h)1.42(t,j=7.50hz,3h)
[0638]
化合物72的制备
[0639][0640]
相应地,化合物72以与化合物73相同的方式制备,从6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体bs开始,以给出0.035g,28%。
[0641]
1h nmr(400mhz,cdcl3)δppm 9.52(s,1h)7.53(d,j=8.38hz,3h)7.29(dd,j=9.48,1.98hz,1h)7.23(d,j=8.38hz,2h)6.86(d,j=8.82hz,2h)6.46(d,j=8.38hz,2h)5.99(br.s.,1h)4.64-4.70(m,1h)4.58(d,j=5.29hz,2h)3.95(s,2h)3.90(s,2h)2.94(q,j=7.50hz,2h)2.80(ddd,j=10.47,6.95,2.87hz,2h)2.43(ddd,j=10.25,6.73,3.31hz,2h)1.38(t,j=7.50hz,3h)
[0642]
化合物74的制备
[0643][0644]
相应地,从中间体bo和中间体bs开始以与化合物73相同的方式制备化合物74,给出0.064g,70%。
[0645]
化合物75的合成
[0646][0647]
中间体bt的制备
[0648]
在schlenck反应器中,将6-boc-2,6-二氮杂螺[3.4]辛烷(cas[885270-86-0],0.5g,2.36mmol)、1-溴-4-(三氟甲氧基)苯(cas[407-14-7],525μl,3.53mmol)和叔丁醇钠(0.453g,4.71mmol)在1,4-二噁烷(25ml)中的溶液用n2吹扫。然后添加乙酸钯(ii)(52.9mg,0.236mmol)和xantphos(0.136g,0.236mmol),再次用n2吹扫混合物并在100℃下搅拌2小时。将混合物合并,通过垫过滤。将滤饼用etoac洗涤,并且将滤液在真空中蒸发以给出1.2g棕色固体。将该残余物通过制备型lc(不规则sioh,15-40μm,50g merck,干法填充流动相梯度:从庚烷/etoac从95/5至60/40)进行纯化,以给出0.756g的呈灰白色固体的中间体bt(80%)。
[0649]
中间体bu的制备
[0650]
向中间体bt(0.706g,1.90mmol)在ch2cl2(20ml)中的溶液中添加三氟乙酸(7.25ml,94.7mmol)(反应混合物变成棕色)并且将混合物在室温下搅拌20分钟。将混合物倾倒入饱和的nahco3溶液中。将各层分离并将水层用ch2cl2萃取。将合并的有机层经mgso4干燥,过滤出并在真空中蒸发以给出棕色油状物,将该油状物在et2o中研磨并且过滤出以给出0.519g呈灰白色粉末(98%)的中间体bu。
[0651]
中间体bv的制备
[0652]
在密封的管中,将中间体bu(0.5g,1.84mmol)、4-溴苄腈(cas[623-00-7],0.5g,2.76mmol)和叔丁醇钠(0.53g,5.51mmol)在1,4-二噁烷(20ml)中的溶液用n2吹扫。然后添加乙酸钯(ii)(0.041 g,0.184mmol)和xantphos(0.106g,0.184mmol),再次用n2吹扫混合物并在100℃下搅拌3小时。将混合物冷却至室温,用垫过滤,并且将滤饼用etoac洗涤。将滤液在真空中蒸发以给出棕色油。将该残余物通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充流动相梯度:从庚烷/etoac从95/5至50/50)进行纯化,以
给出0.429g的黄色油状物(将其静置结晶)。通过反相(固定相:ymc-actus triart-c18 10μm 30x150mm,流动相:从(水性nh4hco30.2%)/can从50/50至0/100的梯度)纯化油以给出0.328g呈黄色固体(48%)的中间体bv。
[0653]
中间体bw的制备
[0654]
在高压釜中,向中间体bv(0.28g,0.75mmol)于氨7m(在甲醇中)(7.8ml)中的溶液中添加拉尼镍并且将混合物在室温下在2巴下氢化1小时。将混合物在垫过滤并且将滤饼用meoh洗涤。将滤液在真空中蒸发以给出黑色固体,将其溶解在etoac中,滤出,并且蒸发滤液以给出0.244g呈白色固体(86%)的中间体bw。
[0655]
化合物75的制备
[0656]
将6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.155g,0.647mmol)、中间体bw(0.244g,0.647mmol)、hatu(0.271g,0.712mmol)和二异丙基乙胺(0.286ml,1.68mmol)在dmf(6.5ml)中的溶液在室温下搅拌过夜。将该混合物在50℃下加热2小时。将该混合物冷却至室温并且在真空中蒸发以给出980mg的黑色油。将该残余物通过制备型lc(不规则sioh,15-40μm,50g,merck,干法填充流动相梯度:从庚烷/etoac从95/5至50/50)进行纯化,以给出0.254g的呈黄色固体的残余物。通过反相(球形c18,25μm,40g ymc-ods-25,干法填充流动相梯度(水性nh4hco30.2%)/mecn从30/70至0/100)纯化残余物以给出白色固体,将该白色固体在戊烷中研磨,过滤并在真空下蒸发(50℃,16小时),给出0.156g呈为白色固体(41%)的化合物75。
[0657]1h nmr(400mhz,dmso-d6)δppm 9.06(s,1h),8.35(br t,j=5.8hz,1h),7.65(d,j=9.6hz,1h),7.44(dd,j=9.6,2.0hz,1h),7.20(br d,j=8.1hz,2h),7.16(br d,j=8.6hz,2h),6.53(br d,j=8.6hz,2h),6.49(br d,j=8.6hz,2h),4.40(d,j=6.1hz,2h),3.82(s,4h),3.46(s,2h),3.25-3.29(m,2h),2.96(q,j=7.4hz,2h),2.23(t,j=6.8hz,2h),1.25(t,j=7.6hz,3h)
[0658]
化合物76的合成
[0659][0660]
中间体bx的制备
[0661]
相应地,中间体bx以与中间体bt相同的方式制备,从6-boc-2,6-二氮杂螺[3.4]辛
烷cas[885270-86-0]和4-溴苄腈cas[623-00-7]开始,给出0.673g,84%。
[0662]
中间体by的制备
[0663]
相应地,中间体by以与中间体bu相同的方式制备,从中间体bx开始,给出0.312g,80%。
[0664]
中间体bz的制备
[0665]
相应地,中间体bz以与中间体bv相同的方式制备,从中间体by和1-溴-4-(三氟甲氧基)苯cas[407-14-7]开始,给出0.369g,73%。
[0666]
中间体ca的制备
[0667]
相应地,中间体ca以与中间体bw相同的方式制备,从中间体bz开始,给出0.2g,56%。
[0668]
化合物76的制备
[0669]
相应地,化合物76以与化合物75相同的方式制备,从6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体ca开始,以给出0.078g,30%。
[0670]1h nmr(500mhz,dmso-d6)δppm 9.07(s,1h),8.41(t,j=6.2hz,1h),7.67(d,j=9.5hz,1h),7.46(dd,j=9.6,1.7hz,1h),7.21(m,j=8.2hz,2h),7.15(br d,j=8.8hz,2h),6.57(d,j=9.1hz,2h),6.46(m,j=8.2hz,2h),4.42(d,j=5.6hz,2h),3.79(s,4h),3.47(s,2h),3.30-3.33(m,2h),2.97(q,j=7.4hz,2h),2.24(t,j=7.0 hz,2h),1.26(t,j=7.4hz,3h)
[0671]
化合物77的合成
[0672][0673]
在n2下,将中间体af(0.1g,0.574mmol)、中间体m(0.28g,0.631mmol)、x-phos(0.033g,0.069mmol)、pd(dba)2(0.02g,0.034mmol)和叔丁醇钠(0.221g,2.30mmol)在二噁烷(4ml)中的溶液在微波下在100℃下辐射1小时。将混合物浓缩。将粗产物经gemini通过高效液相层析法进行纯化(洗脱液:nh3水/乙腈45/55至45/55)。收集所需级分并浓缩,以给出化合物77,0.0076g,3%。
[0674]
1h nmr(400mhz,氯仿-d)δppm 9.53(d,j=1.25hz,1h)8.47(s,2h)7.50-7.56(m,2h)7.30(d,j=2.01hz,1h)7.28(d,j=2.01hz,1h)7.25(s,1h)7.23(s,1h)6.47(d,j==8.53hz,2h)5.99(s,1h)4.59(d,j==5.27hz,2h)4.04(s,2h)3.83(s,2h)3.50(t,j==8.78hz,1h)2.95(q,j==7.53hz,2h)2.63-2.75(m,2h)2.30-2.44(m,2h)1.39(t,j=7.65hz,3h)
[0675]
化合物78的合成
[0676][0677]
中间体cb的制备
[0678]
在n2流中,将2-氟-6-氮杂螺[3.3]庚烷(cas[1354953-09-5],0.8g,6.95mmol)、4-溴苄腈(cas[623-00-7],1.265g,6.95mmol)、binap(0.26g,0.42mmol)、pd2(dba)3(0.127g,0.14mmol)、叔丁醇钠(2g,20.84mmol)和三乙胺(0.48ml)在甲苯(50ml)中的溶液在110℃下搅拌过夜。将混合物浓缩。将残余物溶解于ch2cl2(300ml)和水(150ml)中。将有机层用盐水(150ml)洗涤,经硫酸镁干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(洗脱液:乙酸乙酯/石油醚为从0至1/5)进行纯化。将所希望的级分收集并浓缩以给出1g,66%的中间体cb。
[0679]
中间体cc的制备
[0680]
在25℃(h2,15psi)下,将中间体cb(0.45g,2.08mmol)于氨7m(在meoh中)(20ml)中的混合物用拉尼镍(40mg)作为催化剂氢化16小时。在吸收了h2之后,将催化剂过滤出,并且将滤液进行浓缩以给出中间体cc,0.45g,98%。
[0681]
化合物78的制备
[0682]
在25℃下,将中间体bo(0.048g,0.23mmol)、hatu(0.112g,0.3mmol)、二异丙基乙胺(0.076g,059mmol)在dmf(10ml)中的溶液搅拌30分钟。将中间体cc(0.05g,0.23mmol)添加至混合物中并且将混合物在25℃下搅拌2小时。经菲罗门gemini,通过高效液相层析法(洗脱液:0.05%氨水/甲醇30/70至0/100)纯化粗产物。收集所需级分并冻干,以给出化合物78,0.0134g,14%。
[0683]
1h nmr(400mhz,氯仿-d)δppm 7.18(d,j=8.53hz,2h)6.38-6.45(m,2h)5.83(br.s.,1h)4.86-5.09(m,1h)4.48(d,j=5.52hz,2h)4.31(t,j=5.65hz,2h)3.88(s,2h)3.84(s,2h)3.64(s,2h)2.79(t,j=5.52hz,2h)2.67-2.71(m,2h)2.60-2.67(m,2h)2.38-2.50(m,5h)1.22(t,j=7.53hz,3h)
[0684]
化合物79的合成
[0685][0686]
中间体cd的制备
[0687]
在n2流下,将5-氯-3-碘吡啶-2-胺(cas[211308-81-5],4g,15.72mmol)、2,4-己二酮(cas[3002-24-2],4.50g,34.58mmol)、碳酸铯(5.12g,15.71mmol)、binol(900.20mg,3.14mmol)和碘化亚铜(299.39mg,1.57mmol)在dmso(50ml)中的混合物搅拌15小时。将盐水和乙酸乙酯添加至该混合物中。将有机层分离,用盐水洗涤,经mgso4干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(洗脱液:乙酸乙酯/己烷从0至1/1)进行纯化。将所希望的级分收集并浓缩以给出2.5g,67%的中间体cd。
[0688]
中间体ce的制备
[0689]
在0℃下将氢化钠(0.354g,8.85mmol)添加至中间体cd(2.2g;7.38mmol)在thf(40ml)中的溶液中。搅拌30分钟后,添加碘甲烷(1.26g,8.85mmol)。将混合物升温至25℃并搅拌3小时。将混合物倾倒在冰水中。将混合物用乙酸乙酯萃取(50mlx2)。将有机层合并,用盐水洗涤,经mgso4干燥并过滤。将滤液浓缩。将该粗产物经硅胶柱层析法(洗脱液:乙酸乙酯/石油醚为从0至1/3)进行纯化。将滤液浓缩以给出1.6g,86%的中间体ce。
[0690]
中间体cf的制备
[0691]
在80℃下,将中间体ce(1.6g,6.33mmol)在氢氧化钠水性(5g,62.51mmol,50%在h2o中)溶液中的混合物搅拌过夜。薄层层析法(洗脱液:乙酸乙酯/石油醚=1/3)显示起始材料被消耗。将混合物浓缩。将该混合物用甲基叔丁基醚处理(25mlx2)。将水层用溶液(乙酸乙酯/石油醚=1/3)(2
×
50ml)萃取。用1n hcl调节水层直至ph为4。将残余物过滤并浓缩以给出中间体cf,1.3g,86%。
[0692]
化合物79的制备
[0693]
在25℃下,将中间体cf(0.06g,0.25mmol)、hatu(0.123g,0.33mmol)、二异丙基乙胺(0.08g,0.62mmol)在dmf(10ml)中的溶液搅拌30分钟。将中间体q(0.1g,0.28mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。将粗产物经gemini通过高效液相层析法纯化(洗脱液:0.05%氨/甲醇40/60至10/90)。收集所需级分并浓缩,以给出化合物79,0.052g,36%。
[0694]
1h nmr(400mhz,氯仿-d)δppm 8.22(d,j=1.76hz,1h)7.91(d,j=1.76hz,1h)7.28(s,2h)7.08(d,j=8.82hz,2h)6.49(d,j=8.38hz,2h)6.42(d,j=8.82hz,2h)5.89(br.s.,1h)4.59(d,j=5.29hz,2h)4.04(d,j=2.21hz,7h)3.83(s,3h)3.21(q,j=7.50hz,2h)1.33(t,j=7.72hz,3h)
[0695]
化合物80的合成
[0696][0697]
在25℃下,将5-氯-2-乙基-1-甲基吲哚-3-甲酸(cas[1784796-04-8],0.131g,0.55mmol)、hatu(0.272g,0.72mmol)、二异丙基乙胺(0.185g,1.43mmol)在dmf(10ml)中的溶液搅拌30分钟。将中间体q(0.1g,0.28mmol)添加到混合物中,并将混合物在25℃下搅拌2小时。将混合物在真空下浓缩。经沃特斯xbridge prep obd c18 150x30x5μ通过高效液相层析法(洗脱液:nh3水/乙腈70/65至40/95)纯化残余物。收集所需级分并冻干,以给出化合
物80,0.0423g,13%。
[0698]
1h nmr(400mhz,氯仿-d)δppm 7.62(d,j=1.76hz,1h)7.28(d,j=8.53hz,2h)7.22-7.25(m,1h)7.14-7.19(m,1h)7.08(d,j=8.78hz,2h)6.49(d,j=8.53hz,2h)6.42(d,j=9.03hz,2h)6.01(br.s.,1h)4.61(d,j=5.52hz,2h)4.04(s,8h)3.72(s,3h)3.19(q,j=7.19hz,2h)1.30(t,j=7.53hz,3h)
[0699]
化合物81的合成
[0700][0701]
在n2下,将中间体m(0.1g,0.23mmol)、2-氟-7-氮杂-螺[3.5]壬烷(cas[1263178-15-9],0.049g,0.23mmol)、x-phos(0.0105g,0.022mmol)、pd(dba)2(0.0065g,0.011mmol)和叔丁醇钠(0.055g,0.57mmol)在二噁烷(3ml)中的溶液在100℃下辐射1小时。将混合物浓缩。将粗产物经gemini通过高效液相层析法进行纯化(c18 150x25mmx10μ,25ml/min,洗脱液:nh3水/乙腈45/55至45/55)。收集所需级分并浓缩,以给出化合物81,0.0073g,7%。
[0702]1h nmr(400mhz,cdcl3)δ9.53(d,j=1.5hz,1h),7.54(d,j=9.5hz,1h),7.33-7.26(m,3h),6.95(d,j=8.6hz,2h),6.02(br.s.,1h),5.86(tdd,j=7.4,10.0,17.1hz,1h),5.20-5.08(m,2h),4.61(d,j=5.5hz,2h),3.50(d,j=12.3hz,2h),3.10(dt,j=2.4,12.2hz,2h),2.96(q,j=7.5hz,2h),2.47-2.34(m,2h),1.99-1.63(m,4h),1.39(t,j=7.5hz,3h)
[0703]
化合物82的合成
[0704][0705]
中间体cg的制备
[0706]
将2-氨基吡啶(cas[504-29-0],4.0g;42.5mmol)在thf(220ml)中的溶液冷却至5℃,然后添加丙酰乙酸乙酯(cas[4949-44-4],6.1ml;42.5mmol)、二乙酸碘苯(cas[3240-34-4],13.7g;42.5mmol)和bf3·
oet2(556μl;2.13mmol)。允许所得的混合物加温至室温然后在室温下搅拌过夜。将混合物倾倒进nahco3饱和水性溶液中并用etoac萃取。将合并的有机层用盐水洗涤,经mgso4干燥,过滤并浓缩以给出18.8g呈橙色的固体。将该粗产物吸收在et2o中,从而导致沉淀。过滤沉淀以给出3.8g呈灰白色固体(41%)的粗产物。通过制备型lc(常规二氧化硅30μm,25g,液体填充(ch2cl2),流动相梯度:从庚烷/etoac100/0至50/50)纯化滤液以得到1.7g呈灰白色固体的中间体30,将中间体30吸收在et2o中,过滤固体并在高真空下干燥以得到1.2g呈白色固体(13%)的中间体cg。
[0707]
中间体ch的制备
[0708]
在添加氧化铂(125mg,0.55mmol)和hcl(125μl,1.50mmol)之前,通过n2鼓泡10分钟将中间体cg(1.2g;5.50mmol)在meoh(27ml)中的溶液脱气。将所得的混合物在室温下在1巴下过夜氢化。添加etoac,并且将混合物通过垫过滤,将滤液浓缩至干燥以给出1.4g呈无色油状(定量)的中间体ch。
[0709]
中间体ci的制备
[0710]
将氢氧化锂一水合物(170mg;4.05mmol)添加至中间体ch(300mg;1.35mmol)在meoh(3ml)和h2o(158μl)中的溶液中。将所得混合物在50℃下搅拌48小时。将溶剂在真空中蒸发至干燥以给出与甲苯共沸(2次)的灰白色胶状物,然后在高真空下干燥以给出0.353g呈灰白色固体的中间体ci(原样用于下一步骤中)。
[0711]
化合物82的制备
[0712]
将二异丙基乙胺(0.232ml;1.35mmol)和hatu(0.267g;0.70mmol)依次添加至中间体ci(0.108g;0.54mmol)在dmf(10ml)中的溶液中。将所得混合物在室温下搅拌30分钟,之后添加在dmf(7ml)中的中间体v(0.196g;0.54mmol)。将该混合物在室温下搅拌4小时。将反应混合物在真空中蒸发至干燥,然后用etoac稀释并用盐水洗涤(两次)。经mgso4干燥有机层,过滤并蒸发至干燥以给出585mg的棕色油状物,将其通过制备型lc(常规二氧化硅30μm,12g,干法填充流动相梯度庚烷/etoac/meoh从90/8/2至50/40/10)纯化以得到0.131g的灰白色固体。将固体在et2o中研磨,过滤并在高真空下干燥以给出97mg呈白色固体(经2步33%)的化合物82。
[0713]1h nmr(400mhz,dmso-d6)δppm8.08(t,j=6.1hz,1h)7.35(d,j=8.6hz,2h)7.29(d,j=8.1hz,2h)7.11(d,j=8.6hz,2h)6.39(d,j=8.1hz,2h)4.28(d,j=6.1hz,2h)3.96(t,j=5.6hz,2h)3.91(s,2h)3.70(s,2h)3.47(quint,j=8.8hz,1h)2.66-2.72(m,2h)2.52-2.62(m,4h)2.26-2.34(m,2h)1.74-1.87(m,4h)1.08(t,j=7.3hz,3h)
[0714]
化合物83的合成
[0715][0716]
将二异丙基乙胺(0.31ml,1.78mmol)和hatu(0.353g,0.927mmol)依次添加至6-乙
基-2-甲基咪唑并[2,1-b]噻唑-5-甲酸(cas[1131613-58-5],0.15g,0.713mmol)在dmf(20ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体v(259mg,0.713mmol),并将混合物在室温下搅拌过夜。将该反应混合物用etoac稀释,并且用nahco3饱和水溶液(两次)以及盐水(两次)洗涤。将合并的有机相经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则二氧化硅15-40μm,12g,干法填充(二氧化硅),流动相梯度:从庚烷/etoac 90/10至50/50)纯化粗产物,并且将所得固体在戊烷中研磨,过滤并在45℃下真空干燥以得到0.167g呈白色固体(42%)的化合物83。
[0717]1h nmr(400mhz,dmso-d6)δppm8.01(t,j=5.8hz,1h)7.87(s,1h)7.36(d,j=8.6hz,2h)7.28(d,j=8.6hz,2h)7.15(d,j=8.1hz,2h)6.40(d,j=8.6hz,2h)4.35(d,j=5.6hz,2h)3.91(s,2h)3.70(s,2h)3.47(br t,j=8.6hz,1h)2.84(q,j=7.2hz,2h)2.55-2.62(m,2h)2.41(s,3h)2.25-2.35(m,2h)1.19(t,j=7.58hz,3h)。
[0718]
化合物84和化合物85的合成
[0719][0720]
中间体cj的制备
[0721]
用功率输出范围从0至400w的单模式微波(biotage initiator60),将6-甲氧基-2-氮杂螺[3.3]庚烷盐酸化物(cas[1638761-19-9],0.47g,2.36mmol)、4-氟苯甲腈(cas[1194-02-1],0.576g,4.71mmol)和碳酸钾(0.976g,7.07mmol)在dmso(11ml)中的悬浮液在120℃下加热30分钟[固定保持时间]。将反应混合物在genevac仪器中蒸发并通过制备型lc(不规则二氧化硅,15-40μm,50g,干法填充流动相梯度庚烷/etoac从95/5至70/30)纯化以给出0.361g呈白色固体(67%)的中间体cj。
[0722]
中间体ck的制备
[0723]
在高压釜中,将拉尼镍(0.8g,13.6mmol)添加至中间体cj(0.713g,3.12mmol)于氨7n(在meoh中)(15ml)中的溶液中并且将混合物在室温下在3巴的h2下搅拌过夜。经过滤混合物并真空蒸发以给出0.717g呈蓝色油状物(99%)的中间体ck。
[0724]
化合物84的制备
[0725]
将二异丙基乙胺(0.461ml,2.71mmol)和hatu(436mg,1.15mmol)依次添加至6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.25g,1.04mmol)在dmf(10ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体ck(0.242g,1.04mmol)在dmf(5ml)中的溶液,并且将混合物在室温下搅拌1小时。将该反应混合物在真空中蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,120g,干法填充(二氧化硅),流动相梯度:从dcm100%、meoh0%至dcm 90%、meoh 10%,在20cv中)纯化粗产物以给出0.5g的橙色固体,将其在et2o、et2o/etoh(9:1)、ipr2o和etoh中依次研磨以给出0.317g呈浅橙色固体(69%)的化合物84。
[0726]1h nmr(400mhz,dmso-d6)δppm 9.05(d,j=1.5hz,1h),8.37(t,j=5.8hz,1h),7.65(d,j=9.6hz,1h),7.44(dd,j=9.6,2.0hz,1h),7.17(d,j=8.6hz,2h),6.37(d,j=8.6hz,2h),4.39(d,j=6.1hz,2h),3.75(s,2h),3.81-3.74(m,1h),3.70(s,2h),3.11(s,3h),2.95(q,j=7.4hz,2h),2.47-2.41(m,2h),2.02(ddd,j=10.0,7.0,2.8hz,2h),1.27-1.21(m,3h)
[0727]
化合物85的制备
[0728]
在添加pd/c(0.0032g;3.01μmol)之前,将化合物84(0.08g;114mmol)在meoh(3.5ml)中的溶液通过n2鼓泡脱气5分钟。将所得的混合物在室温下在3巴下过夜氢化。将混合物通过垫过滤,并将滤液在真空下蒸发至干燥。通过制备型lc(常规二氧化硅15-40μm,12g,干法填充流动相梯度:从ch2cl2/meoh 100/0至95/5)纯化粗产物以获得0.057g的固体,将其在庚烷中研磨,过滤并在50℃高真空下干燥持续72小时以给出0.043g呈白色固体(58%)的化合物85。
[0729]1h nmr(500mhz,dmso-d6)δppm 8.19(br s,1h)7.10(d,j=8.2hz,2h)6.36(d,j=8.2hz,2h)4.28(d,j=5.9hz,2h)3.98(br t,j=5.5hz,2h)3.73-3.79(m,3h)3.70(s,2h)3.12(s,3h)2.72(br t,j=5.9hz,2h)2.58-2.65(m,2h)2.41-2.48(m,2h)2.03(m,2h)1.85(br d,j=4.7hz,2h)1.79(br d,j=5.4hz,2h)1.09(t,j=7.6hz,3h)。
[0730]
化合物86的合成
[0731][0732]
将二异丙基乙胺(0.293ml,1.73mmol)和hatu(0.402g,1.06mmol)依次添加至中间体l(0.2g,0.704mmol)在dmf(5ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体ck(0.135g,0.581mmol)在dmf(2.3ml)中的溶液,并且将混合物在室温下搅拌1小时。将该反应混合物在真空中蒸发至干燥以给出0.96g的棕色油状物。通过制备型lc(不规则二氧化硅15-40μm,40g,干法填充流动相梯度:从dcm 99.5%、meoh/水性nh3(95:5)0.5%至dcm 94%、meoh/水性nh3(95∶5)6%)纯化粗产物以得到呈橙色胶状物的0.516g。将产物通过反相(球形c18二氧化硅,25μm,120g ymc-ods-25,干法填充流动相梯度:从60%水溶液(nh4hco30.2%)、40%mecn到20%水溶液(nh4hco30.2%)、80%mecn)纯化以给出0.164g的淡黄色固体,将其在et2o中研磨,过滤并在高真空下干燥以给出0.085g呈白色固体(27%)的化合物86。
[0733]1h nmr(400mhz,dmso-d6)δppm 9.38(d,j=2.5hz,1h),8.67(d,j=2.5hz,1h),8.47(t,j=5.8hz,1h),7.18(d,j=8.6hz,2h),6.37(d,j=8.1hz,2h),4.40(d,j=5.6hz,2h),3.79-3.69(m,5h),3.11(s,3h),2.99(q,j=7.6hz,2h),2.46-2.39(m,2h),2.06-1.97(m,2h),1.26(t,j=7.6hz,3h)
[0734]
化合物87的合成
[0735][0736]
中间体cl的制备
[0737]
在n2下,在经火焰干燥的圆底烧瓶中,将iprmgcl.licl 1.3m(7.14ml,9.28mmol)的溶液添加到4-溴-2-氟苯甲醚(cas[2357-52-0],1.90g,9.28mmol)在无水thf(30ml)中的溶液中。将溶液在室温下在n2流下搅拌5小时,然后在n2下,在0℃下,逐滴添加(约15分钟)至中间体r(1.00g,3.09mmol)、n1,n1,n2,n2-四甲基环己烷-1,2-二胺(cas[38383-49-2],0.063g,0.37mmol)和氯化钴ii(0.04g,0.31mmol)在无水thf(30ml)中的溶液中。将所得混合物在室温下搅拌过周末,用水性nh4cl 10%(40ml)水解并用乙酸乙酯(2
×
40ml)萃取。将合并的有机相经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,220g,干法填充(二氧化硅),流动相梯度:从庚烷/etoac从90/10至60/40)纯化粗产物以给出0.619g呈白色固体(62%)的中间体cl。
[0738]
中间体cm的制备
[0739]
在n2下,将三甲基氯硅烷(1.21ml,9.60mmol)逐滴添加到中间体cl(0.615g,1.91mmol)在无水甲醇(20ml)中的溶液中。将反应混合物在室温下搅拌过夜,并且然后蒸发至干燥以给出0.447g呈白色固体(91%)的中间体cm。
[0740]
中间体cn的制备
[0741]
用功率输出范围从0至400w的单模式微波(biotage initiator60),将中间体cm(0.425g,1.65mmol)、4-氟苯甲腈(cas[1194-02-1],0.3g,2.47mmol)和碳酸钾(0.912g,6.60mmol)在无水dmso(10ml)中的混合物在120℃下加热1小时[固定保持时间]。将该反应混合物用水(40ml)淬灭并且用乙酸乙酯(2
×
50ml)进行萃取。将合并的有机相用水(2
×
50ml)和盐水(2
×
50ml)洗涤,经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,120g,干法填充(二氧化硅),流动相梯度:从庚烷/etoac从90/10至40/60)纯化粗产物以给出0.349g呈白色固体(65%)的中间体cn。
[0742]
中间体co的制备
[0743]
在高压釜中,将中间体cn(0.34g,1.06mmol)和拉尼镍(0.269g,4.58mmol)于氨7n
(在meoh中)(11ml)中的混合物在室温下,在3巴h2下搅拌过夜。然后将反应混合物通过垫过滤并蒸发至干燥以给出0.3g呈灰白色固体(87%)的中间体co。
[0744]
化合物87的制备
[0745]
将二异丙基乙胺(0.19ml,1.09mmol)和hatu(0.175g,0.46mmol)依次添加至6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.1g,0.42mmol)在dmf(7ml)中的溶液中。将所得混合物在室温下搅拌1小时,然后添加中间体co(0.15g,0.46mmol),并且将混合物在室温下搅拌2小时。将该反应混合物在真空中蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,40g,液体填充,流动相梯度dcm/meoh从100/0至95/5)纯化粗产物以给出黄色固体。将该固体在et2o中研磨以给出0.132g黄色固体,将其溶于etoh中并蒸发至干燥以给出0.123g呈浅黄色固体(55%)的化合物87。
[0746]1h nmr(500mhz,dmso-d6)δppm 9.05(s,1h),8.39(br t,j=5.7hz,1h),7.66(d,j=9.5hz,1h),7.45(br d,j=9.5hz,1h),7.19(d,j=8.2hz,2h),7.13-7.04(m,2h),6.99(br d,j=8.5hz,1h),6.41(d,j=8.2hz,2h),4.40(br d,j=5.7hz,2h),3.90(s,2h),3.80(s,3h),3.70(s,2h),3.40-3.33(m,1h),2.96(q,j=7.4hz,2h),2.28-2.23(m,2h),1.25(t,j=7.4hz,3h)
[0747]
化合物88的合成
[0748][0749]
将二异丙基乙胺(0.171ml,1.01mmol)和hatu(0.162g,0.43mmol)依次添加至中间体l(0.11g,0.39mmol)在dmf(4ml)中的溶液中。将所得混合物在室温下搅拌45分钟,然后添加中间体38(0.139g,0.43mmol)在dmf(2ml)中的溶液,并且将混合物在室温下搅拌1小时。将该反应混合物在真空中蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,40g,grace,液体填充,流动相梯度dcm/meoh从100/0至90/10)纯化粗产物以给出褐色固体,将其在et2o中研磨并在50℃高真空下干燥过夜以给出0.098g淡黄色固体。将该固体溶于乙醇中并蒸发至干燥以给出淡黄色固体,将其在ipr2o中研磨以给出0.091g呈白色固体(44%)的化合物88。
[0750]1h nmr(400mhz,dmso-d6)δppm9.39(s,1h),8.67(s,1h),8.47(br s,1h),7.19(d,j=8.1hz,2h),7.13-7.03(m,2h),7.02-6.96(m,1h),6.40(d,j=8.1hz,2h),4.41(br d,j=5.6hz,2h),3.90(s,2h),3.80(s,3h),3.69(s,2h),3.41-3.33(m,1h),3.00(q,j=7.2hz,2h),2.27-2.21(m,2h),1.26(br t,j=7.6hz,3h)
[0751]
化合物89的合成
[0752][0753]
中间体cp的制备
[0754]
将吡啶-4-硼酸(cas[1692-15-5],0.571g,4.64mmol)、钾双(三甲基硅烷基)酰胺(1.14g,6.19mmol)、碘化镍ii(0.097g,0.31mmol)和反式-2-氨基环己醇盐酸化物(cas[5456-63-3],0.036g,0.31mmol)在iproh(20ml)中的溶液在n2下在室温下搅拌5分钟。然后,添加中间体r(1.00g,3.09mmol)并将反应混合物在90℃下加热20小时。将该反应混合物用水(50ml)水解并且用乙酸乙酯(2
×
50ml)进行萃取。将这些有机相进行合并并且用盐水(50ml)洗涤,经mgso4干燥,并且蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,120g,液体填充,流动相梯度dcm/meoh从95/5至90/10)纯化粗产物以给出0.251g呈白色固体(30%)的中间体39。
[0755]
中间体cq的制备
[0756]
在n2下,将三甲基氯硅烷(0.52ml,4.15mmol)逐滴添加到中间体cp(0.227g,0.83mmol)在无水甲醇(10ml)中的溶液中。将反应混合物在室温下搅拌过夜,并且然后蒸发至干燥以给出0.194g呈白色固体(定量)的中间体cq,将其原样用于下一个步骤中。
[0757]
中间体cr的制备
[0758]
用功率输出范围从0至400w的单模式微波(biotage initiator60),将中间体cq(0.179g)、4-氟苯甲腈(cas[1194-02-1],0.206g,1.70mmol)和碳酸钾(0.587g,4.25mmol)在无水dmso(5.5ml)中的混合物在120℃下加热1小时[固定保持时间]。将该反应混合物用水淬灭并且用乙酸乙酯(2次)进行萃取。将合并的有机相用水(2次)和盐水(2次)洗涤,经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,40g,液体填充,流动相梯度dcm/meoh从100/0至95/5)纯化粗产物以给出0.095g呈白色固体(41%)的中间体cr。
[0759]
中间体cs的制备
[0760]
将中间体cr(0.095g,0.35mmol)和拉尼镍(0.088g,1.5mmol)于氨7n(在meoh中)(4ml)中的混合物在室温下,在3巴h2下搅拌过夜。然后将反应混合物通过垫过滤并
蒸发至干燥以给出0.078g呈白色固体(81%)的中间体cs。
[0761]
化合物89的制备
[0762]
将二异丙基乙胺(0.118ml,0.69mmol)和hatu(0.112g,0.29mmol)依次添加至6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.064g,0.27mmol)在dmf(3ml)中的溶液中。将所得混合物在室温下搅拌45分钟,然后添加中间体cs(0.078g,0.28mmol)在dmf(2ml)中的溶液,并且将混合物在室温下搅拌1小时。将该反应混合物在真空中蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,40g,液体填充,流动相梯度dcm/meoh从100/0至90/10)纯化粗产物以给出粘性固体。将该固体在et2o中研磨,然后溶解于dcm中并用水洗涤两次,经mgso4干燥,过滤并蒸发至干燥以给出0.072g的白色固体。将该固体溶于乙醇并蒸发至干燥,然后依次在et2o和ipr2o/etoh(9:1)中研磨。将所得的固体通过制备型lc(球形c18二氧化硅,25μm,40g ymc-ods-25,干法填充流动相梯度:0.2%水性(nh4hco3)/mecn从30∶70至0∶100在6cv中)纯化,并且最后在et2o中研磨以给出0.032g呈白色固体(25%)的化合物89。
[0763]1h nmr(400mhz,dmso-d6)δppm 9.06(s,1h),8.47(br d,j=5.6hz,2h),8.37(br t,j=5.6hz,1h),7.66(d,j=9.6hz,1h),7.44(br d,j=9.6hz,1h),7.25(d,j=4.8hz,2h),7.19(d,j=8.0hz,2h),6.41(br d,j=8.1hz,2h),4.41(br d,j=5.6hz,2h),3.92(s,2h),3.71(s,2h),3.48-3.43(m,1h),2.96(q,j=7.6hz,2h),2.62-2.57(m,2h),2.35-2.29(m,2h),1.25(br t,j=7.3hz,3h)
[0764]
化合物90的合成
[0765][0766]
中间体ct的制备
[0767]
将2-boc-2,6-二氮杂螺[3.3]庚烷草酸酯(cas[1041026-71-4],2.0g,6.73mmol)、4-溴苄腈(cas[623-00-7],1.84g,10.1mmol)和叔丁醇钠(2.59g,26.9mmol)在1,4-二噁烷(70ml)中的溶液脱气。然后添加乙酸钯(0.151g,0.673mmol)和xantphos(0.389g,
0.673mmol),再次用n2吹扫混合物并在100℃下搅拌3小时。将混合物冷却至室温并通过垫过滤。将滤饼用etoac洗涤并将滤液在真空中蒸发。通过制备型lc(不规则二氧化硅,15-40μm,120g,干法填充流动相梯度庚烷/etoac从95/5至60/40)纯化粗产物以给出0.919g呈白色固体(48%)的中间体ct。
[0768]
中间体cu的制备
[0769]
将中间体ct(0.5g,1.67mmol)在甲酸(5ml)中的混合物在室温下搅拌16小时。将混合物在真空中蒸发以给出0.526g呈橙色胶状物的中间体44,将其静置结晶(定量)。
[0770]
中间体cv的制备
[0771]
向中间体cu(0.25mg,0.715mmol)和三乙胺(0.5ml,3.60mmol)在cm(7.5ml)中的溶液在0℃下添加至乙酸酐(0.075ml,0.79mmol)中并且将混合物在0℃下搅拌2小时。将该混合物用dcm稀释并且用水洗涤。将有机层经mgso4干燥,过滤出并在真空中蒸发。通过制备型lc(不规则二氧化硅,15-40μm,24g,液体填充(dcm),流动相梯度dcm/meoh从99/1至94/6)纯化粗产物以给出0.167g呈白色固体(97%)的中间体cv。
[0772]
中间体cw的制备
[0773]
在高压釜中,向中间体cv(0.167g,0.69mmol)于氨7n(在meoh中)(4ml)中的溶液中添加拉尼镍(0.2g,3.4mmol)并且将混合物在室温下在3巴的h2下搅拌2小时。将混合物过滤出并在真空中蒸发以给出0.153g呈白色固体(90%)的中间体cw。
[0774]
化合物90的制备
[0775]
将三乙胺(0.29ml,2.09mmol)和hatu(0.285g,0.75mmol)依次添加至6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.163g,0.68mmol)在dmf(4ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体cw(0.178g,0.726mmol)在dmf(3ml)中的溶液,并且将混合物在室温下搅拌3小时。将该反应混合物在真空中蒸发至干燥以给出0.717g浅黄色固体。通过制备型lc(不规则二氧化硅15-40μm,50g,干法填充流动相梯度dcm/meoh从99/1%至95/5)纯化粗产物以获得0.351g的黄色胶状物。通过反相(固定相:ymc-actus triart-c18 10μm30x150mm,流动相:梯度从70%水性(nh4hco
3 0.2%)、30%mecn至100%mecn)纯化产物以给出0.234g白色固体,将其在et2o中研磨,过滤并在高真空下干燥以给出0.222g呈白色固体(72%)的化合物90。
[0776]1h nmr(400mhz,dmso-d6)δppm 9.06(s,1h),8.41(br s,1h),7.67(d,j=9.5hz,1h),7.46(d,j=9.0hz,1h),7.20(d,j=8.2hz,2h),6.43(d,j=8.2hz,2h),4.41(br d,j=5.0hz,2h),4.28(s,2h),4.00(s,2h),3.91(s,4h),2.96(q,j=7.4hz,2h),1.75(s,3h),1.25(t,j=7.6hz,3h)
[0777]
化合物91的合成
[0778][0779]
中间体cx的制备
[0780]
在0℃下,向中间体cu(0.25g,0.715mmol)和三乙胺(0.50ml,3.60mmol)在dcm(7.5ml)中的溶液中添加苯甲酰氯(0.09ml,0.78mmol)并且将混合物在0℃下搅拌2小时。将该混合物用dcm稀释并且用水洗涤。将有机层经mgso4干燥,过滤出并在真空中蒸发。通过制备型lc(不规则二氧化硅,15-40μm,24g,grace,液体填充(dcm),流动相梯度:从dcm99%、meoh 1%至dcm 96%、meoh 4%)以给出0.128g呈白色固体(59%)的中间体cx。
[0781]
中间体cy的制备
[0782]
在高压釜中,向中间体cx(0.128g,0.422mmol)于氨7n(在meoh中)(2.4ml)中的溶液中添加拉尼镍(0.12g,2.1mmol)并且将混合物在室温下在3巴下搅拌2小时。将混合物过滤出并且在真空下蒸发以给出0.108g呈无色油状的中间体cy,将其静置结晶(83%)。
[0783]
化合物91的制备
[0784]
将二异丙基乙胺(0.168ml,0.99mmol)和hatu(0.168g,0.44mmol)依次添加至6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.09g,0.38mmol)在dmf(2.5ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体cy(0.13g,0.42mmol)在dmf(1.7ml)中的溶液,并且将混合物在室温下搅拌3小时。将该反应混合物在真空中蒸发至干燥以给出0.52g橙色胶状物。通过制备型lc(不规则二氧化硅15-40μm,40g,干法填充流动相梯度dcm/meoh从99/1至94/6)纯化粗产物以获得0.137g黄色胶状物。通过反相(固定相:ymc-actus triart-c18 10μm30x150mm,流动相:梯度从60%水性(nh4hco30.2%)、40%mecn至100%mecn)纯化产物以给出0.109g的无色油状物,将其在et2o中研磨,过滤并在高真空下干燥以给出0.095g呈白色固体(49%)的化合物91。
[0785]1h nmr(500mhz,dmso-d6)δppm 9.06(s,1h),8.40(br t,j=5.8hz,1h),7.68-7.62(m,3h),7.54-7.44(m,4h),7.20(d,j=8.2hz,2h),6.43(d,j=8.5hz,2h),4.49(s,2h),4.41(d,j=5.7hz,2h),4.24(s,2h),3.99-3.90(br q,4h),2.96(q,j=7.4hz,2h),1.25(t,j=7.4hz,3h)
[0786]
化合物92的合成
[0787][0788]
中间体cz的制备
[0789]
在高压釜中,向中间体f(1.57g,5.26mmol)于氨7m(在meoh中)(50ml)中的溶液中添加拉尼镍(1.4g,23.9mmol)并且将混合物在室温下在3巴下氢化过周末(3小时后,所有的氢都被消耗。将高压釜再次填充至3巴的h2)。将混合物过滤并且在真空中蒸发。将残余的灰色胶状物溶于etoac中,与siliamets(r)咪唑(1当量w/w)一起搅拌1小时,然后经垫过滤。将滤液真空蒸发以给出1.29g呈白色固体的中间体cz。
[0790]
中间体da的制备
[0791]
向6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.4g,1.67mmol)在二异丙基乙胺(0.74ml,4.35mmol)和dmf(15ml)中的溶液中添加hatu(0.7g,1.84mmol)并且将混合物在室温下搅拌20分钟。添加中间体cz(505mg,1.67mmol),然后将混合物在室温下搅拌1小时。将混合物在真空中蒸发以给出棕色胶状物。将残余物通过制备型lc(不规则sioh,15-40μm,50g,干法填充庚烷/etoac/meoh(9∶1)从85/15至35/65)纯化以给出0.784g呈白色固体(92%)的中间体da。
[0792]
中间体db的制备
[0793]
向中间体da(0.784g,1.54mmol)在meoh(16ml)中的溶液中添加三甲基氯硅烷(1ml,7.92mmol)并且将混合物在室温下搅拌16小时。将混合物在真空中蒸发以给出0.79g呈浅黄色泡沫状的中间体da(将粗产物原样用于下一个步骤中)。
[0794]
化合物92的制备
[0795]
将三氟乙酸酐(0.235ml,1.69mmol)在0℃下添加至中间体db(0.79g,80%,1.54mmol)和三乙胺(1.1ml,7.91mmol)在dcm(9ml)中的溶液中。将反应混合物在0℃下搅拌1小时,然后在室温下搅拌1小时。将反应混合物用nahco3饱和溶液淬灭并且用dcm(两次)萃取。将有机层经mgso4干燥,过滤出,然后在真空中蒸发,得到0.75g的灰白色泡沫。将残余物通过制备型lc(不规则sioh,15-40μm,50g,干法填充庚烷/etoac/meoh(9∶1)从90/10至60/40)纯化以给出0.643g的白色泡沫。
[0796]
通过反相(球形c18,25μm,120g ymc-ods-25,干法填充流动相梯度:
从35%水性(nh4hco30.2%)、65%mecn至100%mecn)纯化残余物并且将纯净的级分直接冷冻干燥。将蓬松固体溶于mecn中,然后在真空中蒸发以给出无色油状物。该油状物在et2o中研磨并在真空中蒸发以给出0.593g呈白色固体(76%经两步)的化合物92。
[0797]1h nmr(400mhz,dmso-d6)δppm 9.07(d,j=2.0hz,1h),8.46(br t,j=5.8hz,1h),7.66(d,j=9.6hz,1h),7.45(dd,j=9.1,2.0hz,1h),7.30(d,j=8.1hz,2h),7.20(d,j=8.1hz,2h),4.56(s,1h),4.49(d,j=5.6hz,2h),4.33(s,1h),4.23(s,1h),4.01(s,1h),3.41-3.33(m,1h),2.99(q,j=7.6hz,2h),2.68-2.55(m,2h),2.33-2.25(m,2h),1.26(t,j=7.6hz,4h)
[0798]
化合物93的合成
[0799][0800]
将中间体l(0.085g,0.299mmol)和hatu(0.17g,0.447mmol)在二异丙基乙胺(0.13ml,0.764mmol)和dmf(2ml)中的溶液在室温下搅拌30分钟。然后,添加在dmf(1.4ml)中的中间体q(0.085g,0.304mmol)并且将混合物在室温下搅拌3小时。将混合物在真空中蒸发,以给出0.543mg的棕色油状物。将残余物通过制备型lc(不规则sioh,15-40μm,30g,干法填充庚烷/etoac/meoh(9:1)从90/10至45/55)纯化以给出0.088g的黄色胶状物(将其静置结晶)。将残余物在et2o/etoh(9:1)中研磨,过滤出并且在真空(50℃,16小时)下干燥以给出0.064g呈淡黄色固体(36%)的化合物93。
[0801]1h nmr(400mhz,dmso-d6)6ppm 9.38(s,1h),8.67(s,1h),8.51(t,j=5.6hz,1h),7.21-7.15(m,4h),6.68(br t,j=7.1hz,1h),6.44(br d,j=8.1hz,4h),4.41(br d,j=5.6hz,2h),3.95(br s,8h),3.00(q,j=7.6hz,2h),1.26(t,j=7.6hz,3h)。
[0802]
化合物94的合成
[0803][0804]
中间体dc的制备
[0805]
相应地,中间体dc以与中间体bv相同的方式制备,从中间体g(0.315g,1.34mmol)和溴苯开始,给出0.245g,66%。
[0806]
中间体dd的制备
[0807]
相应地,中间体dd以与中间体cz相同的方式制备,从中间体dc(0.14g,0.51mmol)开始以给出0.135g,83%。
[0808]
化合物94的制备
[0809]
将中间体l(0.135g,0.475mmol)和hatu(0.27g,0.71mmol)在二异丙基乙胺(200μl,1.18mmol)和dmf(2.5ml)中的溶液在室温下搅拌30分钟。然后,添加在dmf(2.5ml)中的中间体52(0.135g,0.485mmol)并且将混合物在室温下搅拌3小时。将混合物在真空中蒸发,以给出0.848g的棕色油状物。将残余物通过制备型lc(不规则sioh,15-40μm,40g,干法填充庚烷/etoac/meoh(9∶1)从80/20至35/65)纯化以给出0.159g的淡黄色固体。将固体在et2o/etoh(9∶1)中研磨,过滤出并且在真空(50℃,16小时)下干燥以给出0.118g的白色固体。将该固体通过反相(固定相:ymc-actus triart-c18 10μm30x150mm,流动相:梯度从40%水性(nh4hco30.2%)、60%acn至100%acn)纯化,然后在真空(60℃,16小时)下干燥以给出0.076g呈白色固体(29%)的化合物94。
[0810]
1h nmr(400mhz,dmso-d6)δppm9.40(d,j=3.0hz,1h),8.68(d,j=2.5hz,1h),8.57(t,j=5.8hz,1h),7.32(d,j=8.1hz,2h),7.22(d,j=8.1hz,2h),7.15(t,j=7.8hz,2h),6.65(t,j=7.3hz,1h),6.41(d,j=7.6hz,2h),4.51(d,j=5.6hz,2h),3.93(s,2h),3.71(s,2h),3.42(quin,j=8.7hz,1h),3.03(q,j=7.6hz,2h),2.61-2.54(m,2h),2.34-2.23(m,2h),1.28(t,j=7.6hz,3h)。
[0811]
化合物95和化合物96的合成
[0812][0813]
中间体de的制备
[0814]
将2-boc-2-氮杂螺[3.3]庚-6-酮(cas[1181816-12-5],0.5g,2.37mmol)、1,3-丙二醇(0.26ml,3.55mmol)、原甲酸乙酯(0.39ml,2.37mmol)和氯化锆(0.028g,0.118mmol)在无水dcm(10ml)中的溶液在n2下在室温下搅拌2小时。2小时后,添加0.25当量的原甲酸乙酯(0.099ml,0.59mmol)和0.5当量的1,3-丙二醇(0.09ml,1.18mmol)。
[0815]
5小时后,将该反应混合物用水(30ml)淬灭并且用dcm(30ml)萃取。将有机相用水洗涤,经mgso4干燥,过滤并蒸发至干燥以给出0.633g呈无色油状物的中间体de。
[0816]
中间体df的制备
[0817]
相应地,中间体df以与中间体db相同的方式制备,从中间体de(0.63g,2.35mmol)开始给出0.431g,2.1mmol的盐酸化物盐。
[0818]
中间体dg的制备
[0819]
用功率输出范围从0至400w的单模式微波(biotage initiator60),将中间体df(0.426g,2.07mmol)、4-氟苯甲腈(0.376g,3.11mmol)和碳酸钾(0.859g,6.21mmol)在无水dmso(12ml)中的混合物在120℃下加热1小时[固定保持时间]。将该反应混合物用水(30ml)淬灭,用etoac(2x30ml)萃取。将合并的有机相用水(2
×
30ml)和盐水(2
×
20ml)洗涤,经mgso4干燥,过滤并蒸发至干燥以给出绿色固体。将残余物通过制备型lc(不规则sioh,15-40μm,80g,grace,干法填充(二氧化硅),庚烷/etoac从90/10至50/50)纯化以给出0.369g呈白色固体(66%)的中间体dg。
[0820]
中间体dh的制备
[0821]
相应地,中间体dh以与中间体cz相同的方式制备,从中间体dg(0.334g,1.24mmol)开始以给出0.297g,88%。
[0822]
化合物95的制备
[0823]
将6-氯-2-乙基咪唑并[3,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.225g,0.939mmol)和hatu(0.39g,1.03mmol)在三乙胺(0.39ml,2.81mmol)和dmf(6ml)中的溶液在室温下搅拌30分钟。然后,添加在dmf(5ml)中的中间体dh(0.27g,0.984mmol)并且将混合物在室温下搅拌3小时。将混合物在真空中蒸发,以给出1.22g的橙色胶状物。将残余物通过制备型lc(不规则sioh,15-40μm,50g,merck,干法填充庚烷/etoac/meoh(9∶1)从90/10至45/55)纯化以给出0.483g的白色泡沫(96%)。
[0824]
将51mg残余物溶解在mecn中,用戊烷洗涤(两次)并且在真空中蒸发。将残余的无色油状物在et2o中研磨,过滤出并在高真空(50℃,16小时)下干燥以给出43mg呈白色固体的化合物95。
[0825]1h nmr(400mhz,dmso-d6)δppm 9.05(s,1h),8.37(t,j=5.8hz,1h),7.65(d,j=9.6hz,1h),7.44(dd,j=9.6,2.0hz,1h),7.17(d,j=8.1hz,2h),6.39(d,j=8.6hz,2h),4.40(d,j=6.1hz,2h),3.77-3.72(m,8h),2.95(q,j=7.6hz,2h),2.39(s,4h),1.60-1.55(m,2h),1.24(t,j=7.6hz,3h)
[0826]
化合物96的制备
[0827]
用功率输出范围从0至400w的单模式微波(biotage initiator60),将化合物95(0.35g,0.673mmol)和对-甲苯磺酸(0.013g,0.0673mmol)在丙酮(7.5ml)和水(1.8ml)中的溶液在100℃下加热2小时[固定保持时间]。用功率输出范围从0至400w的单模式微波(biotage initiator60)再次在100℃下加热混合物2小时[固定保持时间]。将混合物用etoac稀释,用nahco3饱和水溶液、盐水洗涤,经mgso4干燥,过滤出并且在真空中蒸发以给出0.301g的黄色固体。将残余物通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充庚烷/etoac/meoh(9∶1)从80/20至40/60)纯化以给出0.275g的灰白色固体。将固体在et2o(3次)中研磨,然后在et2o/etoh(9∶1,2次)中研磨并过滤出以给出0.256g白色固体。
[0828]
将固体通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填无庚烷/etoac/meoh(9:1)从90/10至50/50)纯化并且将纯净的级分直接合并以给出0.151g的白
色固体。将固体通过反相(球形c18,25μm,40g ymc-ods-25,干法填充(硅藻土),流动相梯度:从75%水性(nh4hco30.2%)、25%mecn至35%水性(nh4hco30.2%)、65%mecn)纯化并将纯净的级分冷冻干燥以给出0.045g白色固体的化合物96。
[0829]
1h nmr(400mhz,dmso-d6)δppm9.06(d,j=2.0hz,1h),8.39(br s,1h),7.66(d,j=9.6hz,1h),7.44(dd,j=9.6,2.0hz,1h),7.20(d,j=8.08hz,2h),6.45(d,j=8.59hz,2h),4.41(br d,j=4.6hz,2h),3.96(s,4h),3.35-3.29(m,4h),2.96(q,j=7.6hz,2h),1.25(t,j=7.6hz,3h)
[0830]
还根据在此的这些程序来制备以下化合物:
[0831]
[0832]
[0833]
[0834]
[0835]
[0836][0837]
化合物117、化合物130&化合物131的合成
[0838][0839]
中间体di的制备
[0840]
将三氟甲磺酸银(3.6g,14.1mmol)、(2.49g,7.03mmol)、氟化钾(1.09g,18.8mmol)和6-氧-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1147557-97-8],1.00g,4.69mmol)的悬浮液用n2吹扫。然后,添加在thf(7.03ml,14.1mmol)中的etoac(24ml)、2-氟吡啶(1.21ml,14.1mmol)和三氟甲基三甲基硅烷2m,将混合物再次吹扫并且将所得混合物在室温下搅拌3天。然后将该反应混合物经过滤并且蒸发至干燥。通过制备型lc(不规则二氧化硅,15-40μm,50g,merck,干法填充流动相梯度:庚烷/etoac从90/10至50/50)纯化粗产物以给出0.64g呈白色晶体(49%)的中间体di。
[0841]
中间体dj的制备
[0842]
将三甲基氯硅烷(0.9ml,7.12mmol)添加至中间体di(0.4g,1.42mmol)在无水甲醇(9ml)中的溶液中并且将混合物在室温下搅拌过夜。将混合物在真空中蒸发以给出0.308g
呈淡粉色胶状物的中间体dj,将其静置结晶(定量)。
[0843]
中间体dk的制备
[0844]
用功率输出范围从0至400w的单模式微波(biotage initiator60),将中间体dj(0.308g,1.42mmol)、4-氟苯甲腈(cas[1194-02-1],0.346g,2.83mmol)和碳酸钾(0.782g,5.66mmol)在dmso(7ml)中的悬浮液在120℃下加热30分钟[固定保持时间]。将混合物用etoac稀释,用水(3x),盐水(3x)洗涤,经mgso4干燥,过滤并蒸发。通过制备型lc(不规则二氧化硅,15-40μm,24g,grace,干法填充流动相梯度:庚烷/etoac从95/5至60/40)纯化粗产物以给出0.101g呈白色固体(25%)的中间体dk。
[0845]
中间体dl的制备
[0846]
在高压釜中,向中间体dk(0.101g,0.36mmol)于氨7n(在meoh中)(1.8ml)中的溶液中添加拉尼镍(约0.1g,1.7mmol)并且将混合物在室温下在3巴的h2下搅拌2小时。将混合物过滤并且在真空中蒸发。将滤液吸收在etoac中,并经垫过滤。将滤液在真空中蒸发以给出0.09g呈无色油状物(87%)的中间体dl。
[0847]
化合物117的制备
[0848]
将二异丙基乙胺(0.132ml,0.78mmol)和hatu(125mg,0.33mmol)依次添加至6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.072g,0.30mmol)在dmf(2ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体dl(0.09g,0.31mmol)在dmf(1ml)中的溶液,并且将混合物在室温下搅拌1小时。将该反应混合物在真空中蒸发至干燥以给出0.26g的棕色油状物。通过制备型lc(不规则二氧化硅15-40μm,12g grace,干法填充流动相梯度庚烷/etoac/meoh(9:1)从90/10至50/50)纯化粗产物以得到呈黄色固体的0.126g。将产物通过反相(球形c18,25μm,120g ymc-ods-25,干法填充流动相梯度:从30%水性(nh4hco30.2%)、70%mecn至100%mecn)纯化以给出0.107g白色固体,将其在et2o中研磨,过滤并在高真空下干燥以给出0.085g呈白色固体(57%)的化合物117。
[0849]
1h nmr(400mhz,dmso-d6)δppm 9.05(s,1h),8.37(t,j=5.6hz,1h),7.65(d,j=9.6hz,1h),7.44(dd,j=9.6,1.5hz,1h),7.18(d,j=8.6hz,2h),6.38(d,j=8.6hz,2h),4.79(m,j=7.1hz,1h),4.40(d,j=5.6hz,2h),3.78(d,j=11.6hz,4h),2.95(q,j=7.6hz,2h),2.68-2.55(m,2h),2.45-2.38(m,2h),1.24(t,j=7.6hz,3h)
[0850]
化合物130的制备
[0851][0852]
化合物130以与化合物117相同的方式制备,从中间体ci和中间体dl开始。通过制备型lc(常规sioh 30μm,12g因特奇美拉,干法填充流动相梯度庚烷/etoac/meoh从70:25:5至40:50:10的)纯化粗产物以给出0.099g呈灰白色固体(31%)的化合物130。
[0853]
1h nmr(500mhz,dmso-d6)δppm 8.09(t,j=6.0hz,1h),7.11(d,j=8.5hz,2h),
2-乙基咪唑并[1,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.125g,0.54mmol)在dmf(3.2ml)中的溶液中。将所得混合物在室温下搅拌1小时,然后添加中间体dn(0.152g,0.54mmol)在dmf(3.2ml)中的溶液,并且将混合物在室温下搅拌1小时。将该反应混合物蒸发至干燥。将残余物溶于dcm中并用nahco31%(2
×
)、水(2
×
)和盐水洗涤,经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度dcm/meoh从100/0至90/10)纯化粗产物以得到棕色固体,将其在et2o中研磨以给出0.121g呈灰白色固体(46%)的化合物132。
[0866]
1h nmr(400mhz,dmso-d6)δppm 9.07(s,1h),8.48(t,j=5.8hz,1h),8.02(s,1h),7.85(s,1h),7.82(d,j=2.8hz,1h),7.67(d,j=9.6hz,1h),7.46(dd,j=9.6,2.0 hz,1h),7.31(d,j=7.6hz,2h),7.23(d,j=8.1hz,2h),4.50(d,j=6.1hz,2h),4.17(s,2h),3.96(s,2h),3.42(quin,j=8.9hz,1h),2.99(q,j=7.6hz,2h),2.63-2.57(m,2h),2.33-2.27(m,2h),1.27(t,j=7.6hz,3h)
[0867]
化合物125&化合物133的合成
[0868][0869]
中间体do的制备
[0870]
将2,2-二氟-7-氮杂螺[3.5]壬烷盐酸化物(cas[1263181-82-5],0.3g,1.52mmol)、4-溴苄腈(0.414g,2.28mmol)和叔丁醇钠(0.583g,6.07mmol)在1,4-二噁烷(16ml)中的溶液在n2下脱气。然后添加乙酸钯ii(0.034g,0.152mmol)和xantphos(0.088g,0.152mmol),再次用n2吹扫混合物并在120℃下加热过夜。将混合物冷却至室温,并且在垫过滤。将滤饼用etoac洗涤并将滤液在真空中蒸发。通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充(sioh),流动相梯度庚烷/etoac从90/10至50/50)纯化粗制物以得到0.343g呈黄色固体(86%)的中间体do。
[0871]
中间体dp的制备
[0872]
相应地,中间体dp以与中间体dl相同的方式制备,从中间体do开始,产量0.312g,90%。
[0873]
化合物125的制备
[0874]
将二异丙基乙胺(0.21ml,1.20mmol)和hatu(238mg,0.625mmol)依次添加至6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.111g,0.481mmol)在dmf(13ml)中的溶液中。将所得混合物在室温下搅拌30分钟,然后添加中间体dp(0.128g,0.481mmol),并将混合物在室温下搅拌1小时。将该反应混合物用etoac稀释,并且用nahco3饱和水溶液(两次)以及盐水(两次)洗涤。将有机相经mgso4干燥,过滤并蒸发至干燥以得到0.269g。通过制备型lc(不规则sioh 15-40μm,40ggrace resolv,干法填充(sioh),流动相梯度:庚烷/
etoac从90/10至50/50)纯化粗产物以得到呈白棕色固体的0.199g。将残余物溶于etoac中并用1%水性nahco3(2
×
)、水和盐水(2
×
)洗涤,经mgso4干燥,过滤并蒸发以得到0.183g。将其在ipr2o中研磨,过滤并干燥以得到0.146g的白色固体。将其溶于etoh中并蒸发至干燥(3x)并在真空下干燥过夜以得到0.144g呈白色固体(63%)的化合物125。
[0875]
1h nmr(400mhz,dmso-d6)δppm 9.06(d,j=1.5hz,1h),8.40(t,j=5.8hz,1h),7.66(d,j=9.6hz,1h),7.45(dd,j=9.3,2.3hz,1h),7.21(d,j=8.6hz,2h),6.92(d,j=8.6hz,2h),4.42(d,j=6.1hz,2h),3.10-3.07(m,4h),2.96(q,j=7.6hz,2h),2.39(t,j=13.1hz,4h),1.70-1.67(m,4h),1.25(t,j=7.6hz,3h)
[0876]
化合物133的制备
[0877][0878]
化合物133以与化合物125相同的方式制备,从中间体l和中间体dp开始。通过制备型lc(不规则sioh 15-40μm,40g grace,干法填充(二氧化硅),流动相梯度庚烷/(acoet/meoh 9/1)从90/10至60/40;然后球形c18 25μm,40g ymc-ods-25,干法填充流动相梯度:0.2%水性nh4hco3/mecn从65/35至25/75)纯化粗产物以得到0.083g呈白色固体(36%)的化合物133。
[0879]
1h nmr(500mhz,dmso-d6)δppm 9.39(d,j=2.5hz,1h),8.67(d,j=2.5hz,1h),8.51(t,j=5.7hz,1h),7.21(d,j=8.5hz,2h),6.92(d,j=8.8hz,2h),4.42(d,j=5.7hz,2h),3.10-3.06(m,4h),3.00(q.j=7.4hz,2h),2.39(t,j=13.5hz,4h),1.71-1.67(m,4h),1.26(t,j=7.4hz,3h)
[0880]
化合物134的合成
[0881][0882]
中间体dq的制备
[0883]
将乙酸钯(0.107g,117μmol)和xantphos(0.182g,293μmol)添加至中间体g(0.55g,2.34mmol)、5-溴吡啶(0.373g,2.34mmol)和叔丁醇钠(0.676g,7.03mmol)在1,4-二噁烷(8.3ml)中的混合物中。将空气抽空并且用n2回填。将反应混合物在100℃下加热5小时。冷却至室温后,将反应混合物用acoet和dcm稀释,并且经垫过滤。将滤液蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,120g,grace,干法填充(二氧化硅),流动相梯度
dcm/meoh从100/0至95/5)纯化粗混合物以给出0.306g的呈黄色固体(47%)的中间体dq。
[0884]
中间体dr的制备
[0885]
相应地,中间体dr以与中间体dl相同的方式制备,从中间体dq开始,产出0.298g,96%的黄色固体。
[0886]
化合物134的制备
[0887][0888]
化合物134以与化合物132相同的方式制备,从6氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体dr开始。通过制备型lc(不规则sioh,15-40μm,80g,grace,干法填充(二氧化硅),流动相梯度dcm/meoh从100/0至90/10)纯化粗产物以给出0.268g的呈白色固体(56%)的化合物134。
[0889]
1h nmr(500mhz,dmso-d6)δppm 9.08(s,1h),8.53(s,1h),8.48(t,j=5.6hz,1h),8.03(s,2h),7.68(d,j=9.5hz,1h),7.47(dd,j=9.5,1.9hz,1h),7.32(d,j=7.9hz,2h),7.23(d,j=7.88hz,2h),4.51(d,j=5.6hz,2h),4.09(s,2h),3.88(s,2h),3.46-3.36(m,1h),3.00(q,j=7.6hz,2h),2.62-2.58(m,2h),2.33-2.28(m,2h),1.29-1.24(t,j=7.6hz,3h)
[0890]
化合物135&化合物136的合成
[0891][0892]
中间体ds的制备
[0893]
在n2下,在50℃下,将2,2-二氟-2-(氟磺酰基)乙酸(cas[1717-59-5],1.25g,7.03mmol)在乙腈(6ml)中的溶液在1小时30分钟内添加至叔-丁基6-羟基-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1147557-97-8],1.00g,4.69mmol)和碘化亚铜(0.179g,0.94mmol)在
乙腈(12ml)中的溶液中。将反应混合物在50℃下再搅拌30分钟,然后蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,80g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从100/0至70/30)纯化粗产物以给出0.788g呈白色固体(64%)的中间体ds。
[0894]
中间体dt的制备
[0895]
相应地,中间体dt以与中间体dj相同的方式制备,从中间体ds开始,产出0.563g的无色油状物,定量,将其原样使用。
[0896]
中间体du的制备
[0897]
相应地,中间体du以与中间体dk相同的方式制备,从中间体dt开始,产出0.445g,60%的白色固体。
[0898]
中间体dv的制备
[0899]
相应地,中间体dv以与中间体dl相同的方式制备,从中间体du开始,产出0.433g,96%的无色油状物。
[0900]
化合物135的制备
[0901]
化合物135以与化合物132相同的方式制备,从6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体dv开始。通过制备型lc(不规则sioh,15-40μm,80g,grace,干法填充(二氧化硅),流动相梯度:从dcm 100%、meoh 0%至dcm95%、meoh 5%)以给出0.176g呈白色固体(53%)的化合物135。
[0902]
1h nmr(500mhz,dmso-d6)δppm 9.05(s,1h),8.38(br t,j=5.8hz,1h),7.66(d,j=9.5hz,1h),7.45(dd,j=9.5,1.9hz,1h),7.18(d,j=8.2hz,2h),6.62(t,j=76hz,1h),6.38(d,j=8.5hz,2h),4.54(quin,j=7.2hz,1h),4.40(d,j=6.0 hz,2h),3.78(s,2h),3.74(s,2h),2.95(q,j=7.6hz,2h),2.59-2.55(m,2h),2.30-2.26(m,2h),2.28,1.24(t,j=7.4hz,3h)
[0903]
化合物136的制备
[0904][0905]
化合物136以与化合物135相同的方式制备,从中间体l和中间体dv开始。通过制备型lc(不规则sioh,15-40μm,80g,grace,干法填充(二氧化硅),流动相梯度dcm/meoh从100/0至90/10)纯化粗产物以给出0.127g的呈白色固体(38%)的化合物136。
[0906]
1h nmr(400mhz,dmso-d6)δppm 9.38(d,j=2.5hz,1h),8.67(d,j=2.5hz,1h),8.48(t,j=5.8hz,1h),7.18(d,j=8.6hz,2h),6.62(t,j=76hz,1h),6.37(t,j=8.4hz,2h),4.54(t,j=7.1hz,1h),4.40(d,j=6.1hz,2h),3.78(s,2h),3.74(s,2h),2.99(q,j=7.4hz,2h),2.59-2.54(m,2h),2.30-2.25(m,2h),1.26(t,j=7.6hz,3h)
[0907]
化合物137的合成
[0908][0909]
中间体dw的制备
[0910]
相应地,将中间体dw以与中间体dq相同的方式制备,从中间体g和4-溴二氟甲氧基苯cas[5905-69-1]开始,产出0.13g的白色固体(30%)。
[0911]
中间体dx的制备
[0912]
相应地,中间体dx以与中间体dr相同的方式制备,从中间体dw开始,产出0.264g的白色固体(90%)。
[0913]
化合物137的制备
[0914]
化合物137以与化合物132相同的方式制备,从6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体dx开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度dcm/meoh从100/0至90/10)纯化粗产物以给出0.132g的呈白色固体(69%)的化合物137。
[0915]
1h nmr(400mhz,dmso-d6)δppm9.07(d,j=1.5hz,1h),8.47(t,j=6.1hz,1h),7.67(d,j=9.6hz,1h),7.46(dd,j=9.6,2.0hz,1h),7.31(d,j=8.0hz,2h),7.22(d,j=8.0hz,2h),6.98(t,j=76hz,1h),6.98(d,j=7.9hz,2h),6.43(d,j=7.9hz,2h),4.50(d,j=5.6hz,2h),3.92(s,2h),3.71(s,2h),3.46-3.36(m,1h),2.99(q,j=7.6hz,2h),2.59-2.54(m,2h),2.30-2.25(m,2h),1.27(t,j=7.6hz,3h)
[0916]
化合物138&化合物139的合成
[0917][0918]
中间体dy的制备
[0919]
将2-戊炔酸乙酯(18ml,135mmol)添加至1-氨基碘化吡啶(25g,113mmol)和碳酸钾(19g,135mmol)在dmf(250ml)中的溶液中。将所得混合物在室温下搅拌48小时并且蒸发至干燥。将该残余物溶解在etoac中并用盐水洗涤(3x)。将有机层经mgso4干燥,过滤并蒸发至干燥以给出14.5g棕色固体,将其依次在et2o和mecn中研磨,过滤以给出8.1g呈灰白色固体的中间体dy。将滤液蒸发至干燥并通过制备型lc(常规sioh 30μm,120g因特奇美拉,干法填充流动相梯度庚烷/etoac从100/0至70/30)纯化以给出额外的1.2g呈白色固体的中间体73(总产率:38%)。
[0920]
中间体dz的制备
[0921]
将氢氧化钠水溶液8m(20ml,164mmol)添加到中间体dy(7g,32.1mmol)在thf(39ml)和甲醇(39ml)中的溶液中。将所得混合物在70℃下搅拌过夜。添加hcl(1m)至混合物中,以使该混合物至大约ph7-8。将所得的沉淀过滤并在高真空下干燥以给出5.3g呈灰白色固体(87%)的中间体dz。
[0922]
化合物138的制备
[0923]
向中间体dy(0.1g,0.52mmol)和三乙胺(0.188ml,1.36mmol)在dcm(6ml)中的溶液中添加edci(0.152g,0.78mmol)和hobt(0.108g,0.78mmol)。将所得混合物在室温下搅拌30分钟,之后添加中间体i(0.202g,0.56mmol),然后在室温下搅拌4小时。用水(2x)洗涤该反应混合物。将有机层经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(常规sioh 30μm,25g因特奇美拉,干法填充流动相:庚烷/acoet/meoh 100:35∶5;然后球形c18 25μm,40g ymc-ods-25,液体填充(meoh/mecn),流动相梯度:0.2%水性nh4hco3/mecn从50:50至0:100,然后100%mecn)纯化粗产物以给出白色固体,在et2o中进一步研磨以给出0.145g呈白色固体(52%)的化合物138。
[0924]
1h nmr(500mhz,dmso-d6)δppm8.68(d,j=6.9hz,1h),8.18(t,j=6.0hz,1h),7.87(d,j=8.8hz,1h),7.40-7.36(m,1h),7.30(d,j=8.2hz,2h),7.21(d,j=7.9hz,2h),7.14(d,j=8.5hz,2h),6.95(td,j=6.9,1.3hz,1h),6.45(d,j=7.8hz,2h),4.45(d,j=6.0hz,2h),3.96(s,2h),3.75(s,2h),3.45-3.35(m,1h),3.01(q,j=7.6hz,2h),2.59-2.55(m,2h),2.31-2.25(m,2h),1.25(t,j=7.6hz,3h)
[0925]
化合物139的制备
[0926]
将化合物138(0.1g,0.187mmol)溶解在乙醇(1.3ml)中并且用pd/c 10%(10mg)处理。将反应在大气压力下在60℃在h2下搅拌16小时。将反应混合物经过滤反并用etoac冲洗。将溶剂在减压下去除。通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从70/30至10/90)纯化粗产物以给出0.065g的呈白色固体(65%)的化合物139。
[0927]
1h nmr(500mhz,dmso-d6)δppm7.69(br t,j=6.0hz,1h),7.36(d,j=8.8hz,2h),7.29(d,j=8.5hz,2h),7.10(d,j=8.2hz,2h),6.38(d,j=8.2hz,2h),4.26(d,j=5.7hz,2h),3.96(br t,j=5.8hz,2h),3.91(s,2h),3.69(s,2h),3.50-3.43(m,1h),2.84(br t,j=6.1hz,2h),2.67(q,j=7.5hz,2h),2.60-2.56(m,2h),2.31-2.27(m,2h),1.93-1.88(m,2h),1.76-1.72(m,2h),1.09(t,j=7.6hz,3h)
[0928]
化合物140、化合物141&化合物142的合成
[0929][0930]
中间体ea的制备
[0931]
将2-丁炔酸乙酯(cas[4341-76-8],6.2ml,54.0mmol)添加至1-氨基碘化吡啶(cas[6295-37-0],10g,45mmol)和碳酸钾(7.5g,54mmol)在dmf(100ml)中的溶液中。将所得混合物在室温下搅拌72h。将混合物蒸发至干燥并且将残余物溶解于etoac中并用盐水(3x)洗涤。将有机层经mgso4干燥,过滤并蒸发至干燥以给出5.1g呈棕色固体(55%)的中间体ea。
[0932]
中间体eb的制备
[0933]
相应地,中间体eb以与中间体dz相同的方式制备,从中间体ea开始,产出3.7g,84%的灰白色固体。
[0934]
中间体ec的制备
[0935]
将中间体eb(0.2g,1.14mmol)、hatu(0.475g,1.25mmol)和二异丙基乙胺(0.47ml,3.41mmol)在dmf(15ml)中的溶液在室温下搅拌30分钟,然后添加在dmf(5ml)中的叔-丁基2-(氨甲基)-7-氮杂螺[3.5]壬烷-7-甲酸酯(cas[1160247-15-3],0.303g,1.19mmol)。将所得混合物在室温下搅拌2小时。将混合物蒸发至干燥,并且残余物溶于etoac中并且用nahhco31%的水溶液(2x)、水(2x)和盐水(2x)洗涤。将有机层经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(常规sioh 30μm,12g因特奇美拉,干法填充流动相梯度:庚烷/etoac/meoh 100:0∶0至70∶25:5;然后球形c18 25μm,40g ymc-ods-25,干法填充流动相梯度:0.2%水性nh4hco3/meoh从50:50至10∶90,然后0.2%水性nh4hco3/meoh 10:90)纯化粗产物,以给出0.318g的呈无色油状物(68%)的中间体ec,为无色油状物。
[0936]
中间体ed的制备
[0937]
在0℃下,将在cpme(0.77ml,2.31mmol)中的hcl 3m添加至中间体ec(0.318g,0.77mmol)在甲醇(6ml)中的溶液中。允许所得的混合物加温至室温过夜。在0℃下,添加额外的在cpme(0.51ml,1.54mmol)中的hcl 3m并且允许混合物温热至室温过夜。将混合物蒸发至干燥以给出0.306g呈白色固体(定量)的中间体ed。
[0938]
化合物140的制备
[0939]
将中间体ed(0.26g,0.745mmol)、4-溴三氟甲氧基苯(0.166ml,1.12mmol)和叔丁醇钠(0.286g,2.98mmol)在1,4-二噁烷(10ml)中的混合物通过n2鼓泡脱气10分钟,然后添加乙酸钯(0.016g,75μmol)和xantphos(0.043g,75μmol)。将所得混合物在100℃下搅拌过夜,然后冷却至室温并通过垫过滤。将滤饼用etoac洗涤并将滤液蒸发至干燥。将该残余物溶解在etoac中并用盐水洗涤(2x)。将有机层经mgso4干燥,过滤,并浓缩至干燥。通过制备lc(不规则sioh 15-40μm,10gbiotage,液体填充(dcm),流动相梯度:从庚烷/etoac/
meoh 80:17∶3至60:35:5;然后球形c18 25μm,40g ymc-ods-25,干法填充流动相梯度:0.2%水性nh4hco3/mecn从70:30至0∶100在10cv中,然后5cv在0.2%水性nh4hco3/mecn 0∶100)纯化粗产物以给出0.062g呈白色固体的化合物140(18%)。
[0940]
1h nmr(500mhz,dmso-d6)δppm8.64(d,j=6.6hz,1h),7.84(d,j=8.8hz,1h),7.60(t,j=5.7hz,1h),7.37(td,j=7.9,1.0hz,1h),7.15(d,j=8.8hz,2h),7.00-6.93(m,3h),3.37-3.30(m,2h),3.17-3.12(m,2h),3.07-3.05(m,2h),2.58-2.50(m,1h),2.54(s,3h),1.93-1.88(m,2h),1.69-1.65(m,2h),1.63-1.54(m,4h)
[0941]
化合物141的制备
[0942][0943]
化合物141以与化合物140相同的方式制备,从中间体eb和中间体i开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从70/30至10/90)纯化粗产物以给出0.056g的呈白色固体(43%)的化合物141。
[0944]
1h nmr(500mhz,dmso-d6)δppm 8.65(d,j=6.9hz,1h),8.08(t,j=6.0 hz,1h),7.91(d,j=9.1hz,1h),7.40-7.37(m,1h),7.30(d,j=8.2hz,2h),7.21(d,j=7.9hz,2h),7.14(d,j=8.5hz,2h),6.97-6.95(m,1h),6.45(d,j=8.8hz,2h),4.46(d,j=5.7hz,2h),3.96(s,2h),3.75(s,2h),3.41(quin,j=8.8hz,1h),2.60-2.55(m,2h),2.57(s,3h),2.30-2.26(m,2h)
[0945]
化合物142的制备
[0946][0947]
化合物142以与化合物140相同的方式制备,从中间体eb和中间体q开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从70/30至10/90)纯化粗产物,产出0.165g的呈白色固体(62%)的化合物142。
[0948]
1h nmr(400mhz,dmso-d6)δppm8.64(d,j=6.6hz,1h),7.98(t,j=5.8hz,1h),7.88(d,j=9.1hz,1h),7.39-7.35(m,1h),7.19(d,j=8.0hz,2h),7.16(d,j=8.0 hz,2h),6.97-6.93(m,1h),6.49(d,j=8.6hz,2h),6.43(d,j=8.6hz,2h),4.37(d,j=5.6hz,2h),4.00(s,4h),3.95(s,4h),2.55(s,3h)
[0949]
化合物143的制备
[0950][0951]
化合物143以与化合物140相同的方式制备,从中间体eb和中间体cc开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从
70/30至10/90)纯化粗产物以给出0.252g的呈白色固体(74%)的化合物143。
[0952]
1h nmr(500mhz,dmso-d6)δppm 8.64(d,j=6.6hz,1h),7.98(br t,j=5.8hz,1h),7.87(d,j=8.8hz,1h),7.39-7.35(m,1h),7.17(d,j=8.5hz,2h),6.96-6.93(m 1h),6.37(d,j=8.5hz,2h),5.01(dquin,j=56,6.5hz,1h),4.35(d,j=6.0hz,2h),3.76(s,2h),3.74(s,2h),2.61-2.58(m,2h),2.54(s,3h),2.39-2.30(m,2h)
[0953]
化合物144&化合物145的合成
[0954][0955]
化合物144的制备
[0956]
化合物144以与化合物138相同的方式制备,从中间体dz和中间体q开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从70/30至10/90)纯化粗产物以给出0.128g的呈白色固体(51%)的化合物144。
[0957]
1h nmr(400mhz,dmso-d6)δppm8.66(d,j=7.1hz,1h),8.06(t,j=5.8hz,1h),7.84(d,j=9.1hz,1h),7.38-7.34(m,1h),7.19(d,j=8.0hz,2h),7.15(d,j=8.0 hz,2h),6.95-6.92(m,1h),6.49(d,j=9.1hz,2h),6.43(d,j=8.1hz,2h),4.37(d,j=6.1hz,2h),4.00(s,4h),3.95(s,4h),2.99(q,j=7.6hz,2h),1.24(t,j=7.6hz,3h)
[0958]
化合物145的制备
[0959]
将化合物144(0.32g,0.56mmol)溶解在乙醇(5ml)中并且用pd/c10%(0.064g,0.060mmol)处理。将反应混合物在3巴h2下在室温下搅拌3天,然后在垫过滤并蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,24g,grace,液体填充(dcm),流动相梯度庚烷/(etoac/meoh)(9∶1)从90/0至20/80;然后球形c18,25μm,40gymc-ods-25,干法填充流动相梯度:从50%(水性nh4hco30.2%)、50%mecn至100%mecn然后mecn 100%)纯化粗产品,以给出0.184g呈白色固体(56%)的化合物145。
[0960]
1h nmr(500mhz,dmso-d6)δppm7.69(t,j=5.8hz,1h),7.16(d,j=8.2hz,2h),7.12(d,j=8.2hz,2h),6.49(d,j=8.8hz,2h),6.41(d,j=8.5hz,2h),4.27(d,j=5.7hz,2h),4.0(s,4h),3.99-3.94(m,2h),3.94(s,4h),2.85(t,j=6.3hz,2h),2.67(q,j=7.6hz,2h),1.94-1.88(m,2h),1.77-1.72(m,2h),1.09(t,j=7.4hz,3h)
[0961]
化合物146&化合物147的合成
[0962][0963]
中间体ee的制备
[0964]
在-70℃下,在n2下,将limmds 1.5m(2.64ml,3.96mmol)逐滴添加到中间体dy(0.721g,3.30mmol)在thf(10ml)中的搅拌溶液中。将反应在-70℃下搅拌2小时,然后逐滴添加在thf(2ml)中的六氯化碳(0.938g,3.96mmol)。将反应在室温下搅拌4小时,并且用水和饱和nh4cl水溶液淬灭。用etoac萃取水相。将有机相经mgso4干燥,过滤并蒸发至干燥以给出0.913g中间体ee(定量),将其原样用于下一步。
[0965]
中间体ef/eg的制备
[0966]
将氢氧化钠8m(2.05ml,16.4mmol)添加至中间体ee(813mg,3.22mmol)在thf(3.9ml)和meoh(3.9ml)中的溶液中,将所得的混合物在70℃下搅拌过夜。添加hcl(1m)至混合物中,以使该混合物至大约ph1。将所得的沉淀过滤并在50℃高真空下干燥以给出中间体ef和eg的0.612g混合物,将其原样用于下一步。
[0967]
化合物146&化合物147的制备
[0968]
化合物146和147以与化合物138相同的方式制备,从中间体ef/eg和中间体i的混合物开始。通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从70/30至10/90)纯化粗产物以给出0.128g(61%)的化合物146和0.037g(17%)的化合物147,两种化合物均为白色固体。
[0969]
化合物146
[0970]
1h nmr(400mhz,dmso-d6)δppm 8.36(t,j=5.8hz,1h),7.87(d,j=8.6hz,1h),7.42-7.38(m,1h),7.31-7.26(m,3h),7.21(d,j=8.1hz,2h),7.14(d,j=8.6hz,2h),6.45(d,j=9.1hz,2h),4.46(d,j=5.6hz,2h),3.96(s,2h),3.75(s,2h),3.43-3.38(m,1h),3.04(q,j=7.6hz,2h),2.60-2.54(m,2h),2.31-2.25(m,2h),1.26(t,j=7.3hz,3h)
[0971]
化合物147
[0972]
1h nmr(400mhz,dmso-d6)δppm 8.14(t,j=6.1hz,1h),7.47(d,j=8.4hz,1h),7.40-7.36(m,1h),7.29(d,j=8.0hz,2h),7.21(d,j=8.0hz,2h),7.14(d,j=8.1hz,2h),6.46-6.44(m,3h),4.44(d,j=5.6hz,2h),4.09(s,3h),3.96(s,2h),3.75(s,2h),3.46-3.38(m,1h),3.00(q,j=7.4hz,2h),2.60-2.54(m,2h),2.31-2.25(m,2h),1.23(t,j=7.6hz,3h)
[0973]
化合物148的合成
[0974][0975]
中间体eh的制备
[0976]
在0℃下,向叔-丁基6-氧-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1181816-12-5],1 g,4.73mmol)在干燥的dcm(50ml)中的溶液中添加ast(1.86ml,14.2mmol),然后将混合物温热至室温并搅拌16小时。添加额外的dast(0.62ml,1当量,4.73mmol),然后将混合物在室温下搅拌3小时。将混合物用饱和nahco3淬灭,然后搅拌10分钟。分离各层并且用dcm(2x)萃取水层。将合并的有机层经mgso4干燥,过滤并蒸发至干燥以给出1.02g呈黄色固体(76%)的中间体eh。
[0977]
中间体ei的制备
[0978]
相应地,中间体83以与中间体dj相同的方式制备,从中间体eh开始,产量呈米色固体的0.764g。
[0979]
中间体ej的制备
[0980]
相应地,中间体ej以与中间体dk和4氟苄腈相同的方式制备,从中间体ei开始,产量0.608g,呈白色固体,74%。
[0981]
中间体ek的制备
[0982]
相应地,中间体ek以与中间体dl相同的方式制备,从中间体ej开始,产量0.559g,呈蓝色固体,74%。
[0983]
化合物148的制备
[0984]
化合物148以与化合物117相同的方式制备(使用三乙胺而不是dipea),从中间体l和中间体ek开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充流动相梯度庚烷/(etoac/meoh)(9:1)从85/15至40/60)纯化粗产物以给出0.285g的呈白色固体(70%)的化合物148。
[0985]
1h nmr(500mhz,dmso-d6)δppm 9.38(d,j=2.5hz,1h),8.67(d,j=2.5hz,1h),8.50(t,j=5.8hz,1h),7.19(d,j=8.2hz,2h),6.42(d,j=8.2hz,2h),4.41(d,j=5.7hz,2h),3.85(s,4h),2.99(q,j=7.5hz,2h),2.84(t,j=12.6hz,4h),1.26(t,j=7.6hz,3h)
[0986]
化合物149的合成
[0987][0988]
中间体el的制备
[0989]
在0℃下,在n2下,将iprmgcl.licl 1.3m(7.14ml,9.28mmol)添加至2-氟-4溴苄腈在无水thf(25ml)中的溶液中。将所得的溶液在n2流下在0℃下搅拌4小时,然后在n2下,在0℃下,将其插管(约30分钟)至中间体r和n1,n1,n2,n2-四甲基环己烷-1,2-二胺(cas[38383-49-2],0.063g,0.37mmol)和cocl2(0.04g,0.31mmol)在无水thf(25ml)中的溶液中。将所得的混合物在室温下搅拌18小时,然后用水淬灭。添加etoac,分离水层并用etoac(2x)萃取。将合并的有机层用盐水洗涤,经mgso4干燥,过滤出并蒸发至干燥。通过制备型lc(常规sioh,30μm,80g,因特奇美拉,干法填充流动相梯度庚烷/etoac从95/5至60/40;然后将球形c18,25μm,120g ymc-ods-25,干法填充流动相梯度:从20%(水性nh4hco30.2%)、80%mecn至100%mecn)纯化粗混合物以给出0.933g的呈白色固体,95%的中间体el。
[0990]
中间体em的制备
[0991]
相应地,中间体em以与中间体dj相同的方式制备,从中间体el开始,产量0.608g,呈白色固体,74%。
[0992]
中间体en的制备
[0993]
相应地,中间体en以与中间体dw相同的方式制备,从中间体em和4-溴三氟甲氧基苯开始,产量0.478g,呈白色固体,44%。
[0994]
中间体eo的制备
[0995]
相应地,中间体eo以与中间体dl相同的方式制备,从中间体en开始,产量0.18g,呈蓝色固体,89%。
[0996]
化合物149的制备
[0997]
化合物149以与化合物131相同的方式制备,从6氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]、和中间体eo开始。通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充流动相梯度庚烷/(etoac/meoh)(9:1)从95/5至50/50;然后球形c18,25μm,40gymc-ods-25,干法填充流动相梯度:从50%(水性nh4hco30.2%)、50%mecn至100%mecn然后100%mecn)纯化粗产品,以给出0.148g呈白色固体(41%)的化合物149。
[0998]
1h nmr(500mhz,dmso-d6)δppm 9.06(d,j=1.6hz,1h),8.46(t,j=5.7hz,1h),7.67(d,j=9.5hz,1h),7.46(dd,j=9.5,2.2hz,1h),7.36(t,j=7.9hz,1h),7.15-7.06(m,4h),6.45(d,j=7.8hz,2h),4.54(br d,j=5.7hz,2h),3.96(s,2h),3.75(s,2h),3.44(quin,j=8.8hz,1h),2.98(q,j=7.6hz,2h),2.59-2.55(m,2h),2.32-2.28(m,2h),1.26(t,j=7.6hz,3h)
[0999]
化合物150的合成
[1000][1001]
中间体ep的制备
[1002]
在0℃下,在n2下,将iprmgcl.lic1 1.3m(10.5ml,13.7mmol)添加至4-溴-3氟苄腈(1.39g,6.96mmol)在无水thf(8ml)中的溶液中。将所得的溶液在n2流下在0℃下搅拌4小时。在n2下,在0℃下,在1小时内将该溶液逐滴添加到中间体r(0.75g,2.32mmol)、fe(acac)3(0.082g,0.23mmol)和tmeda(0.84ml,5.57mm01)在无水thf(15ml)中的溶液中。将所得的混合物在室温下搅拌18小时,然后用nh4cl淬灭。添加etoac和水,分离水层并用etoac萃取。合并的有机层用盐水洗涤,经mgso4干燥,过滤并且蒸发到干燥。通过制备型lc(不规则sioh,15-40μm,50g,merck,干法填充流动相梯度庚烷/etoac从100/0至65/35)纯化粗混合物以给出0.391g呈黄色固体(53%)的中间体ep。
[1003]
中间体eq的制备
[1004]
相应地,中间体eq以与中间体em相同的方式制备,从中间体ep开始,产量0.325g,呈绿色胶状物,定量。
[1005]
中间体er的制备
[1006]
相应地,中间体er以与中间体en相同的方式制备,从中间体eq和4-溴三氟甲氧基苯开始,产量0.385g,呈白色固体,81%。
[1007]
中间体es的制备
[1008]
相应地,中间体es以与中间体eo相同的方式制备,从中间体er开始,产量0.229g,呈灰色固体,59%。
[1009]
化合物150的制备
[1010]
化合物150以与化合物149相同的方式制备,从6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体es开始。通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充流动相梯度庚烷/(etoac/meoh)(9∶1)从95/5至50/50)纯化粗
产物以给出0.289g的呈白色固体(84%)的化合物150。
[1011]
1h nmr(400mhz,dmso-d6)δppm 9.08(s,1h),8.48(t,j=5.8hz,1h),7.67(d,j=9.6hz,1h),7.46(dd,j=9.6,2.0hz,1h),7.33(t,j=7.9hz,1h),7.19-7.11(m,4h),6.45(d,j=8.6hz,2h),4.51(br d,j=5.6hz,2h),3.98(s,2h),3.75(s,2h),3.57(quin,j=8.8hz,1h),3.00(q,j=7.4hz,2h),2.61-2.56(m,2h),2.37-2.31(m,2h),1.27(t,j=7.6hz,3h)
[1012]
化合物151的合成
[1013][1014]
中间体et的制备
[1015]
相应地,将中间体et以与中间体dq相同的方式制备,从中间体g和1-溴-4-(三氟甲硫基)苯cas[33347-1]开始,产量0.37g,呈微红色固体,60%。
[1016]
中间体eu的制备
[1017]
在0℃下,在n2下,将中间体et(0.32g,0.855mmol)逐滴添加至lialh4(0.04g,1.05mmol)在干燥的et2o(8ml)中的悬浮液中。将混合物温热至室温,然后回流3小时,并蒸发至干燥。将残余物吸收在meoh中,并经垫过滤。将滤饼用meoh洗涤,并且将滤液蒸发至干燥以给出0.328g的呈浅黄色固体(定量)的中间体eu。
[1018]
化合物151的制备
[1019]
化合物151以与化合物150相同的方式制备,从6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体eu开始。通过制备型lc(不规则sioh,15-40μm,30g,merck,干法填充流动相梯度庚烷/etoac从90/10至10/90)纯化粗产物以给出0.189g的呈白色固体(41%)的化合物151。
[1020]
1h nmr(500mhz,dmso-d6)δppm 9.08(s,1h),8.47(t,j=5.7hz,1h),7.67(d,j=9.5hz,1h),7.49-7.41(m,3h),7.32(d,j=7.9hz,2h),7.23(d,j=7.9hz,2h),6.48(d,j=8.5hz,2h),4.50(br d,j=5.7hz,2h),4.05(s,2h),3.83(s,2h),3.32(quin,j=8.8hz,1h),2.99(q,j=7.6hz,2h),2.66-2.62(m,2h),2.33-2.27(m,2h),1.27(t,j=7.6hz,3h)
[1021]
化合物152的合成
[1022]
[1023]
中间体ev的制备
[1024]
在0℃下,在n2下,将diad(0.74ml,3.75mmol)在甲苯(5ml)中的溶液添加至6-氧-2-氮杂螺[3.3]庚烷-2-甲酸酯(cas[1147557-97-8],0.8g,3.75mmol)、4-氟苯酚(0.421g,3.75mmol)和三苯基膦(1.48g,5.63mmol)在甲苯(35ml)中的溶液中。允许该反应混合物缓慢升温至室温过夜。添加额外的4-氟苯酚(0.21g,1.88mmol)并且将反应在室温下再搅拌3天。将反应混合物蒸发至干燥,然后溶解在最低量的二乙醚中并冷却至0℃。添加大量过量的庚烷,并且在真空下蒸发所得的混合物,其诱导pph3o的沉淀,将其滤出并用二乙醚洗涤。将滤液蒸发至干燥并通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度:庚烷/etoac从90/10至50/50)纯化以给出1.07g呈黄色固体的中间体ev(未获得纯的,但是如此参与到下一步骤中)。
[1025]
中间体ew的制备
[1026]
将中间体ev(0.945g,3.08mmol)和三甲基氯硅烷(1.95ml,15.4mmol)在无水甲醇(31ml)中的溶液在n2下搅拌过夜。然后将反应混合物蒸发至干燥,并且将残余物在et2o中研磨,过滤并干燥以给出0.543g呈米色固体(85%)的中间体ew。
[1027]
中间体ex的制备
[1028]
在n2下,将中间体ew(0.393g,1.90mmol)、4-溴苄腈(0.518g,2.84mmol)和叔丁醇钠(0.729g,7.59mmol)在1,4-二噁烷(20ml)中的混合物脱气。然后,添加乙酸钯(0.043g,0.190mmol)和xantphos(0.11g,0.190mmol),再次用n2吹扫混合物并在120℃下加热过夜。将混合物冷却至室温,并且在垫过滤。将滤饼用etoac洗涤并将滤液蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,80g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至50/50)纯化粗产物以给出0.24g呈黄色固体(41%)的中间体ex。
[1029]
中间体ey的制备
[1030]
相应地,中间体ey以与中间体es相同的方式制备,从中间体ex开始,产量0.304g,呈白色固体,97%。
[1031]
化合物152的制备
[1032]
化合物152以与化合物131(加热50℃)相同的方式制备,从中间体l和中间体ey开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至10/90)纯化粗产物以给出0.157g的呈黄色固体(67%)的化合物152。
[1033]
1h nmr(500mhz,dmso-d6)δppm 9.38(d,j=2.5hz,1h),8.67(d,j=2.8hz,1h),8.49(t,j=5.8hz,1h),7.19(d,j=8.2hz,2h),7.10(t,j=8.8hz,2h),6.86(dd,j=9.1,4.4hz,2h),6.39(d,j=8.2hz,2h),4.63(quin,j=6.9hz,1h),4.40(d,j=6.0hz,2h),3.84(s,2h),3.76(s,2h),2.99(q,j=7.5hz,2h),2.75-2.72(m,2h),2.26-2.22(m,2h),1.26(t,j=7.6hz,3h)
[1034]
化合物153的合成
[1035][1036]
中间体ez的制备
[1037]
将三氟甲磺酸银(4.79g,18.6mmol)、selectfluor(3.30g,9.32mmol)、氟化钾(1.44g,24.9mmol)和叔-丁基2-羟基-7-氮杂螺[3.5]壬烷7-甲酸酯(cas[240401-28-9],1.50g,6.22mmol)溶解在乙酸乙酯(33ml)中。在n2下,添加2-氟吡啶(1.60ml)和三氟甲基三甲基硅烷2m(9.32ml,18.6mmol)并且将所得的混合物在室温下搅拌40小时。然后将该反应混合物经过滤并且蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,120g,grace,干法填充(二氧化硅),流动相梯度:从庚烷/etoac从90/10至80/20)纯化粗混合物以给出0.855g呈白色固体(44%)的中间体ez。
[1038]
中间体fa的制备
[1039]
将中间体ez(0.853g,2.76mmol)溶于甲醇(21ml)中,并在0℃下用在cpme(4.6ml,13.8mmol)中的hcl 3m处理。然后将反应在室温下搅拌过夜。在减压下除去溶剂以给出0.674g呈白色固体(99%)的中间体fa。
[1040]
中间体fb的制备
[1041]
将中间体fa(0.67g,3.21mmol)、4-氟苯甲腈(0.79g,6.45mmol)和碳酸钾(3.53g,25.5mmol)在dmso(32ml)中的悬浮液在120℃下加热过夜。用水淬灭该反应并且用etoac(3x)进行萃取。将合并的有机相用水(3x)和盐水(2x)洗涤,经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则sioh 15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至70/30)纯化粗产物以给出0.747g呈白色固体(70%)的中间体fb。
[1042]
中间体fc的制备
[1043]
相应地,中间体fc以与中间体es相同的方式制备,从中间体fb开始,产量0.241g,呈白色固体,95%。
[1044]
化合物153的制备
[1045]
化合物153以与化合物152相同的方式制备,从中间体l和中间体103开始。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至10/90)纯化粗产物以给出0.222g的呈黄色固体(59%)的化合物153。
[1046]
1h nmr(500mhz,dmso-d6)δppm 9.39(d,j=2.8hz,1h),8.67(d,j=2.5hz,1h),8.50(t,j=5.8hz,1h),7.21(d,j=8.5hz,2h),6.90(d,j=8.5hz,2h),4.89(quin,j=7.2hz,1h),4.42(d,j=5.7hz,2h),3.09-3.07(m,2h),3.04-3.02(m,2h),3.00(q,j=7.5hz,2h),2.36-2.32(m,2h),1.98-1.94(m,2h),1.66-1.64(m,4h),1.27(t,j=7.4hz,3h)
[1047]
化合物154的合成
[1048][1049]
中间体fd的制备
[1050]
相应地,中间体fd以与中间体es相同的方式制备,从中间体f开始,产量1.29g,呈白色固体,81%。
[1051]
中间体fe的制备
[1052]
向6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸(cas[1216142-18-5],0.117g,0.504mmol)在dcm(5.1ml)和三乙胺(0.18ml)中的溶液中添加edci(145mg,0.756mmol)和hobt(103mg,0.760mmol)并且将混合物在室温下搅拌30分钟。添加中间体fd(0.162g,0.536mmol),并且将混合物在室温下搅拌4小时。用水(2x)洗涤该混合物。将有机层经mgso4干燥,过滤并蒸发至干燥以给出0.293g呈无色油状物的中间体fe(定量),将其原样用于下一步。
[1053]
中间体ff的制备
[1054]
向中间体fe(0.291 g,0.572mmol)在甲醇(5.9ml)中的溶液中添加三甲基氯硅烷(0.37ml,2.94mmol)并且将混合物在室温下搅拌16小时。将混合物蒸发至干燥以给出0.304g呈浅黄色泡沫(定量)的中间体ff。
[1055]
化合物154的制备
[1056]
在0℃下,将三氟甲磺酸酐(0.12ml,0.696mmol)添加至中间体ff(155mg,0.348mmol)和dmap(2.13mg,17.4μmol)在三乙胺(0.39ml,2.78mmol)和dcm(5.3ml)中的溶液中。将所得混合物在0℃下搅拌6小时。添加水并且将有机层用水洗涤,经mgso4干燥,过滤并且蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至10/90)纯化粗产物以给出186mg的浅黄色固体,将其在庚烷中研磨并通过制备型lc(球形c1825μm,40g ymc-ods-25,干法填充流动相梯度:0.2%水性nh4hco3/mecn从90/10至0/100)纯化以给出0.112g呈白色固体(59%)的化合物154。
[1057]
1h nmr(400mhz,dmso-d6)δppm9.07(s,1h),8.47(br s,1h),7.67(d,j=8.1hz,1h),7.46(br d,j=9.1hz,1h),7.30(br d,j=8.1hz,2h),7.20(br d,j=7.6hz,2h),4.49(br d,j=5.1hz,2h),4.41(s,2h),4.18(s,2h),3.39-3.31(m,1h),2.98(q,j=7.4hz,2h),2.63-2.58(m,2h),2.34-2.29(m,2h),1.26(br t,j=7.3hz,3h)
[1058]
化合物155&化合物156的合成
[1059][1060]
中间体fg的制备
[1061]
相应地,中间体fg以与中间体dk相同的方式制备,从2-硫代-6-氮杂螺[3.3]庚烷2,2-二氧化物cas[1263182-09-7]和4氟苯甲腈开始,产量0.206g,呈白色固体,51%。
[1062]
中间体fh的制备
[1063]
相应地,中间体fh以与中间体es相同的方式制备,从中间体fg开始,产量0.208g,呈白色固体,93%。
[1064]
化合物155的制备
[1065]
化合物155以与化合物153相同的方式制备,从6-氯-2-乙基咪唑并[1,2-a]吡啶-3-甲酸cas[1216142-18-5]和中间体fh开始。通过制备型lc(不规则sioh,15-40μm,24g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至10/90)纯化粗产物,产出0.252g的呈白色固体(69%)的化合物155。
[1066]
1h nmr(400mhz,dmso-d6)δppm9.05(s,1h),8.40(br s,1h),7.66(d,j=8.6hz,1h),7.45(d,j=9.6hz,1h),7.21(d,j=7.1hz,2h),6.46(d,j=8.1hz,1h),4.47(d,j=1.5hz,4h),4.41(br s,2h),3.99(s,4h),2.96(q,j=7.4hz,2h),1.25(t,j=7.4hz,3h)
[1067]
化合物156的制备
[1068]
在添加pd/c 10%(8.99mg,8.44μmol)之前,将化合物155(0.117g;255μmol)在甲醇(5.6ml)中的溶液通过n2鼓泡脱气5分钟。将所得的混合物在室温下在5巴h2下搅拌过夜。将混合物通过垫过滤,用etoac冲洗并蒸发至干燥。通过制备型lc(规则sioh 15-40μm,24ggrace,干法填充(二氧化硅),流动相梯度庚烷/etoac从70/30至0/100然后meoh 100%)纯化粗产物以给出0.095g的呈白色固体(69%)的化合物156。
[1069]
1h nmr(400mhz,dmso-d6)δppm 8.10(br t,j=5.6hz,1h),7.13(d,j=8.6hz,2h),6.44(d,j=8.1hz,2h),4.46(s,4h),4.29(br d,j=6.1hz,2h),3.98(s,4h),3.97-3.94(m,2h),2.71-2.68(m,2h),2.58(q,j=7.4hz,2h),1.83-1.78(m,4h),1.08(t,j=7.6hz,3h)
[1070]
化合物157的合成
[1071][1072]
中间体fi的制备
[1073]
将2-氨基-3-氟吡啶(cas[21717-95-3],0.2g,1.78mmo)在me-thf(9ml)中的溶液冷却至5℃。依次添加3-氧代戊酸乙基3-氧代戊酸乙酯(0.50ml,3.53mmol)、二乙酸碘苯(0.578g,1.79mmol)和三氟化硼醚合物(0.024ml,0.089mmol)。将溶液在5℃下搅拌2小时,并且然后升温至室温过夜。添加etoac和饱和nahco3溶液。分离各层并且用etoac萃取水层。将合并的有机层经mgso4干燥,过滤并蒸发至干燥。通过制备型lc(不规则sioh,15-40μm,120g,grace,液体填充(dcm),流动相梯度庚烷/etoac从90/10至70/30)纯化粗混合物以给出0.274g呈白色固体(65%)的中间体fi。
[1074]
中间体fj的制备
[1075]
向中间体fi(0.172g,0.73mmol)在水(2.4ml)和乙醇(2.4ml)中的溶液中添加氢氧化钠(0.088g,2.19mmol)并且将混合物在室温下搅拌过夜。用hcl(3n)将混合物酸化至ph3。蒸发etoh并且将残余物用koh溶液碱化。通过过滤收集所得的白色沉淀,并且将其用hcl(1m)酸化至ph 1,并且将白色固体过滤,干燥以给出0.119g呈白色固体(79%)的中间体fj。
[1076]
化合物157的制备
[1077]
化合物157以与化合物131相同的方式制备,从中间体fj和中间体i开始。通过制备型lc(不规则sioh 15-40μm,40g,grace,干法填充(二氧化硅),流动相梯度庚烷/etoac从90/10至10/90)纯化粗产物以给出0.091g的呈白色固体(42%)的jnj-65053092-aaa。
[1078]
1h nmr(500mhz,dmso-d6)δppm 8.76(d,j=6.9hz,1h),8.56(br t,j=5.7hz,1h),7.32-7.27(m,3h),7.22(d,j=8.2hz,2h),7.14(d,j=8.5hz,2h),7.00-6.96(m,1h),6.45(d,j=9.1hz,2h),4.50(d,j=6.0hz,2h),3.96(s,2h),3.75(s,2h),3.45-3.38(m,1h),2.99(q,j=7.6hz,2h),2.60-2.55(m,2h),2.30-2.26(m,2h),1.27(t,j=7.6hz,3h)
[1079]
还根据在此披露的这些程序来制备以下化合物:
[1080]
化合物158
[1081][1082]
化合物159
[1083][1084]
化合物160
[1085][1086]
化合物161
[1087][1088]
化合物162
[1089][1090]
化合物163
[1091][1092]
化合物164
[1093][1094]
化合物165
[1095][1096]
化合物166
[1097][1098]
化合物167
[1099][1100]
化合物168
[1101][1102]
特征性数据表
[1103]
[1104][1105]
进一步的特征性数据
[1106]
[1107]
[1108]
[1109]
[1110]
[1111]
[1112]
[1113]
[1114]
[1115]
[1116]
[1117]
[1118]
[1119][1120]
分析方法
[1121]
lcms
[1122]
用lcms(液相层析质谱法)记录一些化合物的质量。在下文描述了所使用的方法。
[1123]
通用程序方法c
[1124]
如相应方法中所指明,使用lc泵、二极管阵列(dad)或uv检测器和柱进行高效液相层析(hplc)测量。若需要,包括其他检测器(参见下文的方法表格)。
[1125]
将来自柱的流带至配置有大气压离子源的质谱仪(ms)。设置调谐参数(例如扫描范围、停留时间等)以便获得允许鉴别化合物的标称单一同位素分子量(mw)的离子是在技术人员的知识内。利用适宜软件进行数据采集。通过实验保留时间(r
t
)和离子描述化合物。如果未在数据表中不同地指定,那么报道的分子离子对应于[m+h]
+
(质子化的分子)和/或[m-h]-(去质子的分子)。在该化合物不是直接可电离的情况下,指定加合物类型(即[m+nh4]
+
、[m+hcoo]-等)。对于具有多种同位素模式的分子(br、cl等)来说,报道的值是针对最低同位素质量获得的值。所获得的所有结果均具有实验不确定性,这通常与所使用方法相关。
[1126]
在下文中,“sqd”意指单四极检测器,“rt”意指室温,“beh”意指桥连乙基硅氧烷/二氧化硅杂合物,“hss”意指高强度二氧化硅,“dad”意指二极管阵列检测器。
[1127]
表:lcm方法代码(以ml/min来表示流量;以℃表示柱温度(t);以分钟表示运行时间)。
[1128]
[1129]
通用程序lcms方法a和b
[1130]
如相应方法中所指明,使用lc泵、二极管阵列(dad)或uv检测器和柱进行高效液相层析(hplc)测量。若需要,包括其他检测器(参见下文的方法表格)。
[1131]
将来自柱的流带至配置有大气压离子源的质谱仪(ms)。设置调谐参数(例如扫描范围、停留时间等)以便获得允许鉴别化合物的标称单一同位素分子量(mw)的离子是在技术人员的知识内。利用适宜软件进行数据采集。通过实验保留时间(rt)和离子描述化合物。如果未在数据表中不同地指定,那么报道的分子离子对应于[m+h]
+
(质子化的分子)和/或[m-h]-(去质子的分子)。在该化合物不是直接可电离的情况下,指定加合物类型(即[m+nh4]
+
、[m+hcoo]-等)。对于具有多种同位素模式的分子(br、cl等)来说,报道的值是针对最低同位素质量获得的值。所获得的所有结果均具有实验不确定性,这通常与所使用方法相关。
[1132]
在下文中,“msd”为质谱选择性检测器,“dad”为二极管阵列检测器。
[1133]
表:lcm方法代码(以ml/min来表示流量;以℃表示柱温度(t);以分钟表示运行时间)。
[1134][1135]
当一种化合物是在lcms方法中给出不同峰的异构体的混合物时,仅在lcms表中给出主要成分的保留时间。
[1136]
通用程序
[1137]
如相应方法中所指明,使用lc泵、二极管阵列(dad)或uv检测器和柱进行高效液相层析(hplc)测量。如果必要的话,包括另外的检测器(参见以下方法表)。
[1138]
将来自柱的流带至配置有大气压离子源的质谱仪(ms)。设置调谐参数(例如扫描范围、停留时间等)以便获得允许鉴定化合物的标称单一同位素分子量(mw)的离子在技术人员的知识内。使用适当的软件进行数据采集。通过实验保留时间(rt)和离子描述化合物。如果未在数据表中不同地指定,那么报道的分子离子对应于[m+h]
+
(质子化的分子)和/或[m-h]-(去质子的分子)。在该化合物不是直接可电离的情况下,指定加合物类型(即[m+nh4]
+
、
[m+hcoo]-等)。对于具有多种同位素模式的分子(br、cl等)来说,报道的值是针对最低同位素质量获得的值。所获得的所有结果均具有实验不确定性,这通常与所使用方法相关。
[1139]
在下文中,“sqd”意指单四极检测器,“rt”意指室温,“beh”意指桥连乙基硅氧烷/二氧化硅杂合物,“hss”意指高强度二氧化硅,“dad”意指二极管阵列检测器。
[1140]
表:lcm方法代码(以ml/min来表示流量;以℃表示柱温度(t);以分钟表示运行时间)。
[1141][1142]
在下文中,“msd”为质谱选择性检测器,“dad”为二极管阵列检测器。
[1143]
表:lcm方法代码(以ml/min来表示流量;以℃表示柱温度(t);以分钟表示运行时间)。
[1144]
[1145][1146]
药理学实例测试化合物对抗结核分枝杆菌的mic测定
[1147]
测试1
[1148]
在含有7h9培养基的96孔板中制备实验化合物和参比化合物的适当溶液。从对数生长期的培养物中取出结核分枝杆菌菌株h37rv的样品。首先将它们稀释以在600nm波长处获得0.3的光密度,然后以1/100稀释,从而得到每孔大约5x10exp5集落形成单位的接种物。将板在37℃下于塑料袋中孵化以防止蒸发。7天后,向所有孔中添加刃天青。两天后,在具有543激发和590nm发射波长的gemini em酶标仪上测量荧光,并且计算(或可以计算)mic
50
和/或pic
50
值(等等,例如ic
50
、ic
90
、pic
90
等)。
[1149]
测试2
[1150]
用100μl米德尔布鲁克(middlebrook)(1x)7h9肉汤培养基填充圆底无菌96孔塑料微量滴定板。随后,将额外100μl培养基添加至列2。将化合物的储备溶液(200x最终测试浓度)以2μl体积添加至列2中的一系列复孔中,以便允许评估其对细菌生长的作用。使用多道加样枪(multipipette)在微量滴定板中从列2至列11直接制备连续的2倍稀释液。每3个稀释液后更换移液管吸头,以最小化高疏水化合物的移液误差。在每个微量滴定板中都包括具有(列1)和不具有(列12)接种物的未处理的对照样品。将米德尔布鲁克(1x)7h9肉汤培养基中的100μl体积中的每孔大约10000cfu的结核分枝杆菌(菌株h37rv)添加至行a至h,列12除外。将相同体积的不具有接种物的肉汤培养基添加至列12的行a至h中。将培养物在潮湿气氛中在37℃下孵育7天(具有开放空气阀和连续通风的培养箱)。在第7天,用肉眼检查该细菌生长。
[1151]
将90%最小抑制浓度(mic
90
)确定为无可视化细菌生长的浓度。
[1152]
测试3:时间杀死测定
[1153]
在时间杀死测定中,可以使用肉汤稀释法确定化合物的杀菌或抑菌活性。在关于结核分枝杆菌(菌株h37rv)的时间杀死测定中,结核分枝杆菌的起始接种物在米德尔布鲁克(1x)7h9肉汤中是106cfu/ml。以0.1至10倍mic
90
的浓度使用抗细菌化合物。未接受抗细菌
剂的管构成培养物生长对照。将包含微生物和测试化合物的管在37℃下孵育。在孵育0、1、4、7、14和21天后,取走样品,以通过在米德尔布鲁克7h9培养基中系列稀释(10-1
至10-6
)并在米德尔布鲁克7h11琼脂上铺板(100μl)来确定活菌落计数。将这些板在37℃下孵育21天并且确定菌落的数目。通过每ml的log
10
cfu对时间作图,可以构建杀菌曲线。杀菌效果通常被定义为:与未处理的接种物比较,每ml的cfu数目的3-log
10
减少。通过系列稀释并计数用于铺板的最高稀释度处的菌落来除去药物的潜在的延滞效应(carryover effect)。
[1154]
测试4(也参见上文的测试1;在该测试中使用了不同的结核分枝杆菌菌株)
[1155]
在含有7h9培养基的96孔板中制备实验化合物和参比化合物的适当溶液。从稳定生长期的培养物中取得结核分枝杆菌菌株eh4.0(361.269)的样品。首先将它们稀释以在600nm波长处获得0.3的光密度,然后以1/100稀释,从而得到每孔大约5x10exp5集落形成单位的接种物。将板在37℃下于塑料袋中孵化以防止蒸发。7天后,向所有孔中添加刃天青。两天后,在具有543nm激发和590nm发射波长的gemini em酶标仪上测量荧光,并计算(或可以计算)mic50和/或pic50值(等等,例如ic50、ic90、pic90等)。能以μg/ml在下文记录pic
50
值。
[1156]
结果
[1157]
例如,当在上述的测试1或测试2中进行测试时,本发明/实例的化合物通常可具有从0.01至10μg/ml的ic
90
值。例如,当在上述的测试1或测试2中进行测试时,本发明/实例的化合物通常可具有从3至10(例如从4.0至9.0,例如从5.0至8.0)的pic
50
。
[1158]
在上述的测试1(在“药理学实例”部分)中测试了实例中的化合物,并且获得了以下结果:
[1159]
生物学数据表
[1160][1161][1162]
*和**表示在相关测定中重复的(第二次和第三次)测试;结果中可能会有一定的实验偏差
[1163]
进一步的生物学数据
[1164]
在上述的测试4(在“药理学实例”部分)中测试了实例中的化合物,并且获得了以下结果:
[1165]
[1166]