1.本发明属于聚乳酸催化剂领域,具体涉及到一种催化丙交酯开环聚合的同多钼酸配位聚合物和制备方法。
背景技术:
2.聚乳酸(polylatic acid,简称pla),是一种以乳酸为主要原材料合成的聚酯类聚合物。pla本身无毒,具备良好的生物相容性和生物可吸收性,在生物体内可完全降解为水和二氧化碳,具有非常好的安全性,是公认的理想绿色高分子材料。而且,pla作为少数已被美国食品和药品管理局批准的生物可降解聚合物支架材料,已广泛应用于成骨或成软骨支架及接合材料、皮肤创伤敷料、医用创口缝线、药物缓释及控释载体等医疗领域。
3.合成pla的方法主要有乳酸直接缩聚法和丙交酯开环聚合法两种,直接缩聚法工艺简单,成本较低,得到的分子量相对较低,产物不够纯净,易出现淡黄色或黄棕色的pla,应用的领域也比较窄。
4.目前,相对分子质量高(大于8万)的pla通常都是丙交酯在均相有机金属化合物的催化作用下开环聚合而得。其中催化剂辛酸亚锡应用最广泛,但是包括辛酸亚锡在内的许多有机金属化合物的金属残留物对产品的质量和应用产生不利的影响。在丙交酯开环聚合过程中,采用高活性的非均相催化剂是一种非常好的解决方案,然而到目前为止,相关的文献报道大多集中在非均相催化剂二氧化硅负载金属有机化合物方面(abdel-fattah t.m.,pinnavaia t.j.chem.commun.1996,(5),665-666;yu k.,jones c.w.j.catal.2004,222,558-564;wanna n.,kraithong t.,khamnaen t.,phiriyawirut p.,charoenchaidet s.,tantirungrotechaij.catal.commun.2014,45,118-123),多酸类配位聚合物在丙交酯开环聚合中的研究还未有文献报道。
技术实现要素:
5.本部分的目的在于概述本发明的实施例的一些方面以及简要介绍一些较佳实施例。在本部分以及本技术的说明书摘要和发明名称中可能会做些简化或省略以避免使本部分、说明书摘要和发明名称的目的模糊,而这种简化或省略不能用于限制本发明的范围。
6.鉴于上述和/或现有技术中存在的问题,提出了本发明。
7.因此,本发明的目的是,克服现有技术中的不足,提供一种应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂。
8.为解决上述技术问题,本发明提供了如下技术方案:一种应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂,其化学式为[mo2o4(μ
2-oh)2(htrz)],htrz为1,2,4-三氮唑配体,[mo2o4]
2+
为双核的同多钼酸根阳离子,μ
2-oh为桥连的羟基。
[0009]
作为本发明所述应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的一种优选方案,其中:所述催化剂,其二级结构单元为:
[0010]
晶体属于正交晶系,空间群为pbcm,分子式为c2h5mo2n3o6,分子量为358.99;
[0011]
晶胞参数为:α=90
°
,β=90
°
,γ=90
°
,晶胞体积为z=4;
[0012]
基本结构为一个由[mo2o4]
2+
与1,2,4-三氮唑配体和μ
2-oh配位的二维层状结构。
[0013]
本发明的再一个目的是,克服现有技术中的不足,提供一种应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的制备方法。
[0014]
为解决上述技术问题,本发明提供了如下技术方案:一种应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的制备方法,包括,
[0015]
将二水合钼酸钠、1,2,4-三氮唑、六水合硝酸锌和去离子水加入到高压釜中,于160~190℃反应48~96小时;
[0016]
反应结束后降温到室温,得到所述同多钼酸配位聚合物晶体,再依次经去离子水和乙醇洗涤,干燥制得所述同多钼酸配位聚合物催化剂。
[0017]
作为本发明所述应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的制备方法的一种优选方案,其中:六水合硝酸锌与二水合钼酸钠物质量的比为1:1~1:5。
[0018]
作为本发明所述应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的制备方法的一种优选方案,其中:六水合硝酸锌与1,2,4-三氮唑物质量的比为1:4~1:8。
[0019]
作为本发明所述应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的制备方法的一种优选方案,其中:二水合钼酸钠与1,2,4-三氮唑物质量的比为1:1~1:5。
[0020]
作为本发明所述应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂的制备方法的一种优选方案,其中:每0.6毫摩尔的1,2,4-三氮唑对应添加3~9毫升的水。
[0021]
本发明的另一个目的是,克服现有技术中的不足,提供一种同多钼酸配位聚合物催化剂在催化丙交酯开环聚合制备聚乳酸中的应用,包括,
[0022]
将同多钼酸配位聚合物催化剂和丙交酯加入到干燥的schlenk反应管中,在反应温度下进行本体开环聚合反应,制得聚乳酸;
[0023]
其中,所述的本体开环聚合反应温度为150~190℃,反应时间为6~24小时。
[0024]
作为本发明所述应用的一种优选方案,其中:同多钼酸配位聚合物和丙交酯摩尔比为1:5000~1:30000。
[0025]
作为本发明所述应用的一种优选方案,其中:聚乳酸重均分子量为7万~12万,分子量分布指数为1.3~1.8。
[0026]
本发明有益效果:
[0027]
本发明首次提出一种应用于催化丙交酯开环聚合的同多钼酸配位聚合物催化剂及其制备方法,所制得的同多钼酸配位聚合物热稳定性好,其合成方法简单易行,且产率较高,重现性好;该同多钼酸配位聚合物对丙交酯本体开环具有较好的催化活性,制备的聚乳酸重均分子量超过7万,可以应用在医疗、降解塑料制品和包装材料领域。
附图说明
[0028]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其它
的附图。其中:
[0029]
图1为本发明实施例中同多钼酸配位聚合物中钼酸根的配位环境图。
[0030]
图2为本发明实施例中同多钼酸配位聚合物二维层状结构图。
[0031]
图3为本发明实施例中同多钼酸配位聚合物的粉末衍射示意图。
[0032]
图4为本发明实施例中同多钼酸配位聚合物的热重示意图。
[0033]
图5为本发明实施例中同多钼酸配位聚合物催化丙交酯本体开环聚合转化率与反应时间示意图。
具体实施方式
[0034]
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合说明书实施例对本发明的具体实施方式做详细的说明。
[0035]
在下面的描述中阐述了很多具体细节以便于充分理解本发明,但是本发明还可以采用其他不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似推广,因此本发明不受下面公开的具体实施例的限制。
[0036]
其次,此处所称的“一个实施例”或“实施例”是指可包含于本发明至少一个实现方式中的特定特征、结构或特性。在本说明书中不同地方出现的“在一个实施例中”并非均指同一个实施例,也不是单独的或选择性的与其他实施例互相排斥的实施例。
[0037]
实施例1
[0038]
同多钼酸配位聚合物催化剂的制备:将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率47.4%(25.5毫克,基于mo)。
[0039]
红外数据(kbr,cm
–1):3565s,3376b r,2363w,1596m,1173w,1096m,918s,759s w,539s。
[0040]
实施例2
[0041]
将六水合硝酸锌(59.4毫克,0.2毫摩尔)、1,2,4-三氮唑(29.7毫克,0.1毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(12毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率34.5%(19.4毫克,基于mo)。
[0042]
实施例3
[0043]
将六水合硝酸锌(14.85毫克,0.05毫摩尔)、1,2,4-三氮唑(29.7毫克,0.1毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,在190℃下反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率36.5%(19.8毫克,基于mo)。
[0044]
实施例4
[0045]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(55.2毫克,0.8毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(12毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率37%(20.1毫克,基于mo)。
[0046]
实施例5
[0047]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(27.6毫克,0.4毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(12毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率35.1%(18.9毫克,基于mo)。
[0048]
实施例6
[0049]
同多钼酸配位聚合物催化剂的制备:将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(24.2毫克,0.1毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应48小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率28.9%(15.6毫克,基于mo)。
[0050]
实施例7
[0051]
同多钼酸配位聚合物催化剂的制备:将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(121毫克,0.5毫摩尔)和去离子水(3毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率34.2%(18.4毫克,基于mo)。
[0052]
实施例8
[0053]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(12毫升)加入到15毫升的聚四氟乙烯高压釜中,160℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率41.8%(22.1毫克,基于mo)。
[0054]
实施例9
[0055]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(3毫升)加入到15毫升的聚四氟乙烯高压釜中,190℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率40.1%(21.6毫克,基于mo)。
[0056]
实施例10
[0057]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(12毫升)加入到15毫升的聚四氟乙烯高压釜中,190℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率40.1%(21.6毫克,基于mo)。
[0058]
实施例11
[0059]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(29.7毫克,0.1毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,160℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率37.7%(20.3毫克,基于mo)。
[0060]
实施例12
[0061]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(55.2毫克,0.8毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应96小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率
42.6%(22.9毫克,基于mo)。
[0062]
实施例13
[0063]
将六水合硝酸锌(29.7毫克,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(3毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应48小时后,以5℃/小时的速率缓慢冷却至室温,得到无色块状晶体,产率41.7%(22.4毫克,基于mo)。
[0064]
对比例1
[0065]
将六水合硝酸锌替换成其它锌盐(如乙酸锌、氯化锌或硫酸锌)或其它金属盐(如硝酸铜、硝酸银、硝酸钴、硝酸铁、硝酸钙、硝酸镁、硝酸铝或硝酸镧,0.1毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,160℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,均未获得适合x-射线单晶分析的晶体样品,只得到结构不明确的无定形粉末。
[0066]
对比例2
[0067]
将二水合钼酸钠替换成其它钼酸盐(四水合钼酸铵或磷钼酸,0.3毫摩尔)、1,2,4-三氮唑(41.4毫克,0.6毫摩尔)、六水合硝酸锌(29.7毫克,0.1毫摩尔)和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,均未获得适合x-射线单晶分析的晶体样品,只得到结构不明确的无定形粉末。
[0068]
对比例3
[0069]
将1,2,4-三氮唑替换成其它氮杂环类配体(1-甲基-1,2,4-三氮唑、4-氨基-1,2,4-三氮唑、苯并咪唑或2-甲基咪唑,0.6毫摩尔)、六水合硝酸锌(29.7毫克,0.1毫摩尔)、二水合钼酸钠(72.6毫克,0.3毫摩尔)的和去离子水(6毫升)加入到15毫升的聚四氟乙烯高压釜中,180℃反应72小时后,以5℃/小时的速率缓慢冷却至室温,均未获得适合x-射线单晶分析的晶体样品,只得到结构不明确的无定形粉末。
[0070]
实施例14
[0071]
实施例1制备的同多钼酸配位聚合物催化剂表征
[0072]
(1)同多钼酸配位聚合物催化剂的晶体结构测定
[0073]
晶体结构测定采用brukerapex ii ccd衍射仪,于293(2)k下,用经石墨单色化的mo kα射线以ω扫描方式收集衍射点,收集的数据通过saint程序还原并用sadabs方法进行半经验吸收校正。结构解析和精修分别采用shelxtl程序的shelxs和shelxl完成,通过全矩阵最小二乘方法对f2进行修正得到全部非氢原子的坐标及各向异性参数。所有氢原子在结构精修过程中被理论固定在母原子上,赋予比母原子位移参数稍大(c
–
h,1.2,n
–
h,1.2或o
–
h,1.5倍)的各向同性位移参数。详细的晶体测定数据见表1。
[0074]
表1同多钼酸配位聚合物的主要晶体学数据:
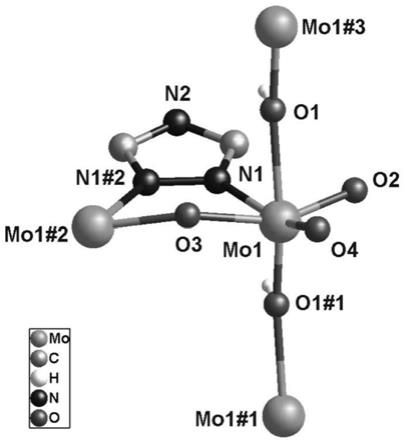
[0075][0076][0077]
钼酸根的配位环境见图1。二维网络结构见图2。图1:同多钼酸配位聚合物中钼酸根的配位环境图。图2:同多钼酸配位聚合物二维层状结构图。
[0078]
(2)同多钼酸配位聚合物的相纯度表征
[0079]
同多钼酸配位聚合物的粉末衍射表征显示其具有可靠的相纯度,为其作为催化丙交酯开环聚合的应用提供了保证。见图3。(仪器型号:rigaku d/max-2500)
[0080]
(3)同多钼酸配位聚合物的热稳定性表征
[0081]
同多钼酸配位聚合物的热稳定性可通过热重分析仪来表征,结果表明,该同多钼酸配位聚合物骨架具有较高的热稳定性,能稳定到380℃。见图4:同多钼酸配位聚合物的热重示意图。(仪器型号:netzsch/tg 209f3)
[0082]
实施例15
[0083]
将实施例1中的同多钼酸金属有机框架材料用于催化丙交酯制备pla。
[0084]
将25.9克丙交酯(0.18摩尔)和4.31毫克同多钼酸配位聚合物(0.012毫摩尔)加入到schlenk反应管中,在160℃下保温反应18小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0085]
经1h nmr定量分析检测丙交酯的转化率与反应时间关系,见图5:含铜同多钼酸金属有机框架材料催化丙交酯开环聚合转化率与反应时间示意图。(仪器型号:brukeravance 400mhz)
[0086]
实施例16
[0087]
将25.9克丙交酯(0.18摩尔)和12.93毫克同多钼酸配位聚合物(0.036毫摩尔)加入到schlenk反应管中,在160℃下保温反应24小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0088]
实施例17
[0089]
将25.9克丙交酯(0.18摩尔)和2.155毫克同多钼酸配位聚合物(0.006毫摩尔)加入到schlenk反应管中,在190℃下保温反应18小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0090]
实施例18
[0091]
将25.9克丙交酯(0.18摩尔)和4.31毫克同多钼酸配位聚合物(0.012毫摩尔)加入到schlenk反应管中,在190℃下保温反应24小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0092]
实施例19
[0093]
将25.9克丙交酯(0.18摩尔)和4.31毫克同多钼酸配位聚合物(0.012毫摩尔)加入到schlenk反应管中,在160℃下保温反应6小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0094]
实施例20
[0095]
将25.9克丙交酯(0.18摩尔)和2.155毫克同多钼酸配位聚合物(0.006毫摩尔)加入到schlenk反应管中,在150℃下保温反应18小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0096]
实施例21
[0097]
将25.9克丙交酯(0.18摩尔)和12.93毫克同多钼酸配位聚合物(0.036毫摩尔)加入到schlenk反应管中,在190℃下保温反应18小时。聚合反应毕,冷却至室温,用30ml二氯甲烷溶解,经离心回收同多钼酸配位聚合物催化剂,再往滤液加入360ml甲醇析出聚合物,于50℃真空中干燥,得到白色产品pla。
[0098]
实施例22
[0099]
(1)实施例15分子量的测定
[0100]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为115290,分子量分布指数为1.44。
[0101]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测
器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正。
[0102]
(2)实施例16分子量的测定
[0103]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为106202,分子量分布指数为1.52。
[0104]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正。
[0105]
(3)实施例17分子量的测定
[0106]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为91235,分子量分布指数为1.62。
[0107]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正。
[0108]
(4)实施例18分子量的测定
[0109]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为80012,分子量分布指数为1.51。
[0110]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正
[0111]
(5)实施例19分子量的测定
[0112]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为103587,分子量分布指数为1.48。
[0113]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正
[0114]
(6)实施例20分子量的测定
[0115]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为66702,分子量分布指数为1.60。
[0116]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正
[0117]
(7)实施例21分子量的测定
[0118]
取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为89402,分子量分布指数为1.49。
[0119]
测试条件:柱温40℃;洗脱液:四氢呋喃;流速0.6毫升/分钟;检测器:rid-10a检测器;校正:使用分子量在2000至250000不等的四种不同标准聚苯乙烯进行分子量校正。
[0120]
对比例4
[0121]
将25.9克丙交酯(0.18摩尔)和六水合硝酸锌(29.7毫克,0.1毫摩尔)加入到
schlenk反应管中,在180℃下保温反应18小时。经1h nmr定量分析检测丙交酯的转化率为45.6%。取7.0毫克pla,溶于1毫升四氢呋喃溶液,经过0.4微米孔径的聚四氟乙烯滤膜过滤,取20微升加入到岛津(日本)制“lc-20ad gpc”进样器中,通过计算得出重均分子量为11402,分子量分布指数为1.76。
[0122]
对比例5
[0123]
将25.9克丙交酯(0.18摩尔)和2.9毫克二水合钼酸钠(0.3毫摩尔)加入到schlenk反应管中,在180℃下保温反应18小时。经1h nmr定量分析检测丙交酯的转化率,发现配体二水合钼酸钠对催化丙交酯开环聚合活性极低,单体转化率为5%,重均分子量小于630,分子量分布指数为1.09。
[0124]
对比例6
[0125]
将25.9克丙交酯(0.18摩尔)和0.8毫克1,2,4-三氮唑(0.6毫摩尔)加入到schlenk反应管中,在180℃下保温反应18小时。经1h nmr定量分析检测,丙交酯没有转化,未能得到pla。
[0126]
本发明多氧钼酸配位聚合物催化材料,不仅结构变化具有多样性,并且细胞毒性低、稳定性高、催化丙交酯性能优异,这将为设计高效、低毒和稳定的丙交酯开环聚合提供重要支持。
[0127]
本发明公开了一种催化丙交酯开环聚合的同多钼酸配位聚合物和制备方法,涉及聚乳酸催化剂领域,其化学式为[mo2o4(μ
2-oh)2(htrz)],htrz为1,2,4-三氮唑配体,[mo2o4]
2+
为双核的同多钼酸根阳离子,μ
2-oh为桥连的羟基。本发明采用二水合钼酸钠和六水合硝酸锌与有机配体1,2,4-三氮唑在封闭条件下,经由水热反应得到具有二维层状结构的同多钼酸配位聚合物。本发明提供的制备方法简单易行,且产率较高,重现性好,所述含同多钼酸配位聚合物对催化丙交酯本体开环聚合具有较好的催化活性,制得的聚乳酸重均分子量超过11万,在医疗、降解塑料制品和包装材料等领域具有较好的应用前景
[0128]
应说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的精神和范围,其均应涵盖在本发明的权利要求范围当中。