1.本发明属于新化合物提取技术领域,具体涉及一种查尔酮衍生物 及其制备和在制备抗肿瘤药物或功能食品中的应用。
背景技术:
2.这里的陈述仅提供与本发明相关的背景技术,而不必然地构成现 有技术。
3.植物通过多种生物合成途径产生具有化学结构多样性的天然产 物,在药物发现过程中发挥着越来越重要的作用。值得注意的是, 1981-2014年间批准的创新药物超过50%来源于天然产物。美国fda 调查结果表明,来源于天然产物的创新药物占批准化学类药物三分之 一以上。
4.查耳酮类化合物是类黄酮家族的主要成分,这类天然产物的特征 是开链黄酮类化合物,具有两个芳环,通过三碳原子连接链包含一个 α,β-不饱和羰基核心单元。历史上,含查耳酮的植物也被用于传统医 学以治疗多种疾病。对这些含有查耳酮的植物进行植物化学研究后, 分离出了众多查耳酮类化合物,一些已用于临床试验,治疗癌症、心 血管疾病和病毒感染。此外,据报道查耳酮类化合物在体外和体内显 示出抗菌、抗原生动物、抗癌、心脏保护、抗糖尿病、神经保护、抗 氧化、抗炎和抗hiv作用。
5.黄毛茛属于毛茛属,广泛分布于巴基斯坦、喜马拉雅地区、印度 和俄罗斯。黄毛茛在传统医学中用于治疗眼部疾病,例如结膜炎及其 伤口愈合。该植物的汁液被用来治疗痛风、间歇性发烧和哮喘,黄毛 茛叶子制成的药物用于治疗关节疼痛和胃气。此外,毛茛属其他植物 广泛的应用于治疗疟疾、瘰疬、痔疮、淋巴结核和关节炎,但是对黄 毛茛中的化合物组成研究较少。
技术实现要素:
6.针对现有技术存在的不足,本发明的目的是提供黄毛茛中新查耳 酮化合物及其提取方法与应用。
7.为了解决以上技术问题,本发明第一方面提供了查耳酮衍生物, 其结构式选自:
[0008][0009]
第二方面,本发明提供上述查耳酮衍生物的制备方法,包括如下 步骤:
[0010]
将干燥黄毛茛进行乙醇回流提取,得粗提物;
[0011]
将粗提物进行硅胶柱色谱层析,采用正己烷-乙酸乙酯洗脱体系 梯度洗脱;
[0012]
将第4个馏分正己烷-乙酸乙酯溶剂系统在硅胶填料上进行二次 色谱分离,得到化合物1和化合物2;
[0013]
将第5个馏分采用正己烷-乙酸乙酯溶剂系统在硅胶填料上进行 二次色谱分离,得到化合物3、化合物4和化合物5。
[0014]
在一些实施例中,采用正己烷-乙酸乙酯洗脱体系梯度洗脱的程 序为正己烷、正己烷-乙酸乙酯、乙酸乙酯洗脱,从0:100到0:100。
[0015]
在一些实施例中,回流提取时,每千克干燥黄毛茛采用3-4l乙 醇回流提取。
[0016]
进一步的,回流提取完毕后,在35-45℃下旋蒸除去溶剂。
[0017]
在一些实施例中,第4个馏分进行二次色谱分离时,正己烷和乙 酸乙酯的体积比为16:84。
[0018]
在一些实施例中,第5个馏分进行二次色谱分离时,正己烷和乙 酸乙酯的体积比为15:85、20:80、25:75梯度洗脱。
[0019]
在一些实施例中,还包括将第2个馏分进行二次色谱分离获得 4-methoxylonchocarpin的步骤。
[0020]
进一步的,进行二次色谱分离时,正己烷与乙酸乙酯的体积比为 5:95、10:90、15:85。分离得到4-methoxylonchocarpin为化合物6。
[0021]
第三方面,本发明提供4-methoxylonchocarpin在制备治疗尤文氏 肉瘤药物中的应用;
[0022]
或,4-methoxylonchocarpin在制备治疗三阴性乳腺癌抗癌药物中 的应用;
[0023]
或,4-methoxylonchocarpin在制备功能食品中的应用。
[0024]
本发明的以上一种或几种实施例取得的有益效果如下:
[0025]
通过以上分离提取方法对黄毛茛中的查尔酮化合物进行提取,得 到5种新的查尔酮化合物和一种已知查尔酮化合物,为黄毛茛的研究 及进一步应用奠定一定基础。
附图说明
[0026]
构成本发明的一部分的说明书附图用来提供对本发明的进一步 理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对 本发明的不当限定。
[0027]
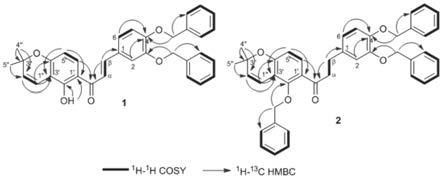
图1是化合物1和化合物2的cosy和hmbc相关图;
[0028]
图2是化合物3、化合物4和化合物5的cosy和hmbc相关 图;
[0029]
图3是化合物6对sk-n-mc(尤文氏肉瘤)、hel(红白血病)、 mcf-7和mda-mb-468(均为乳腺癌)以及pc-3(前列腺癌)活力 影响测试图;
[0030]
图4是化合物a(1)的1h-nmr的核磁共振图;
[0031]
图5是化合物a(1)的
13
c-nmr核磁共振图;
[0032]
图6是化合物a(1)的dept图;
[0033]
图7是化合物a(1)的cosy图;
[0034]
图8是化合物a(1)的hsqc图;
[0035]
图9是化合物a(1)的hmbc图;
[0036]
图10是化合物a(1)的hresims质谱图;
[0037]
图11是化合物b(2)的1h-nmr核磁共振图;
[0038]
图12是化合物b(2)的
13
c-nmr核磁共振图;
[0039]
图13是化合物b(2)的dept图;
[0040]
图14是化合物b(2)的cosy图;
[0041]
图15是化合物b(2)的hsqc图;
[0042]
图16是化合物b(2)的hmbc图;
[0043]
图17是化合物b(2)的hresims质谱图;
[0044]
图18是化合物c(3)的1h-nmr核磁共振图;
[0045]
图19是化合物c(3)的
13
c-nmr核磁共振图;
[0046]
图20是化合物c(3)的cosy图;
[0047]
图21是化合物c(3)的hsqc图;
[0048]
图22是化合物c(3)的hmbc图;
[0049]
图23是化合物c(3)的hresims质谱图;
[0050]
图24是化合物d(4)的1h-nmr核磁共振图;
[0051]
图25是化合物d(4)的
13
c-nmr核磁共振图;
[0052]
图26是化合物d(4)的cosy图;
[0053]
图27是化合物d(4)的hsqc图;
[0054]
图28是化合物d(4)的hmbc图;
[0055]
图29是化合物d(4)的hresims质谱图;
[0056]
图30是化合物e(5)的1h-nmr核磁共振图;
[0057]
图31是化合物e(5)的cosy图;
[0058]
图32是化合物e(5)的hsqc图;
[0059]
图33是化合物e(5)的hmbc图;
[0060]
图34是化合物e(5)的hresims质谱图。
具体实施方式
[0061]
应该指出,以下详细说明都是例示性的,旨在对本发明提供进一 步的说明。除非另有指明,本发明使用的所有技术和科学术语具有与 本发明所属技术领域的普通技术人员通常理解的相同含义。
[0062]
实施例
[0063]
实验方法与试剂
[0064]
所有细胞系均购自atcc(美国马纳萨斯)。用于细胞培养的 rpmi1640、dmem和mem基础细胞培养基、fcs、l-谷氨酰胺、 pbs和胰蛋白酶/edta购自capricorn scientific gmbh(德国 ebsdorfergrund公司)。培养瓶、多孔板和其他细胞培养塑料分别购 自tpp(瑞士trasadingen公司)和greiner bio-one gmbh(德国 frickenhausen公司)。刃天青购自sigma-aldrich chemie gmbh(德 国taufkirchen公司)。
[0065]
实验材料
[0066]
黄毛茛药材于2018年7月从采集于巴基斯坦开伯尔-普赫图赫瓦 省丘陵地区。
[0067]
提取和分离过程
[0068]
干燥黄毛茛药材4.0kg用15l乙醇回流提取,在40℃下旋蒸除 去溶剂,得到5.3g植物提取物。粗提物经过硅胶柱色谱层析(正己 烷、正己烷-乙酸乙酯、乙酸乙酯洗脱,从0:100到0:100),分为九 个组分(f1-9)。
[0069]
馏分f4(200mg)使用溶剂系统正己烷-乙酸乙酯(16:84)在30 g硅胶填料上进行二次色谱分离,得到化合物a(1,4.9mg)和化合 物b(2,4.1mg)。
[0070]
馏分f5(120mg)使用溶剂系统正己烷-乙酸乙酯(15:85至25:75) 在30g硅胶填料上进行二次色谱分离,得到化合物c(3,2.1mg)、 化合物d(4,2.6mg)和化合物e(5,2.4mg)。
[0071]
馏分f2(90mg)使用溶剂系统正己烷-乙酸乙酯(5:95至15:85) 在15g硅胶填料上进行二次色谱分离,得到4-methoxylonchocarpin (6,7mg)。
[0072]
细胞培养
[0073]
使用到的五种人类癌细胞系:pc-3(前列腺癌)、mcf-7(乳腺 癌)、mda-mb-468(三阴性乳腺癌细胞系)、sk-n-mc(尤文氏肉 瘤)和hel(红白血病细胞)。
[0074]
mcf-7、pc-3和hel保存于rpmi 1640培养基中,并补充有 10%的热灭活fcs、2mm的l-谷氨酰胺和1%青霉素/链霉素。 mda-mb-468细胞系加到10%热灭活fcs和1%青霉素/链霉素的 dmem培养基中培养。sk-n-mc细胞加入有10%fcs和1%青霉素 /链霉素的mem培养基中。所有细胞系都在t-75烧瓶中常规培养, 在5%的co2的潮湿环境中,在37℃下达到亚汇合(~80%),再进行 继代培养或测定使用。在细胞传代和接种前,贴壁细胞mcf-7、 mda-mb-468、pc3和sk-n-mc用pbs洗涤并用胰蛋白酶/edta (0.05%溶于pbs)分离。使用前,重新悬浮在标准培养基,收集并 离心hel悬浮细胞(室温800rpm)。
[0075]
体外细胞活力测定
[0076]
采用荧光刃天青细胞活力测定法,分别研究化合物的抗增殖和细 胞毒性作用。将癌细胞接种到96孔板中,贴壁细胞系密度为6000个 细胞/100μl/孔,悬浮细胞系的密度为20,000个细胞/100μl/孔。随后, 将细胞粘附24小时,用化合物处理48小时。
[0077]
制备查耳酮化合物dmso储备溶液(20mm),在上述标准培 养基中稀释,以达到细胞培养所需浓度。
[0078]
对于对照样品,将细胞与0.5%的dmso(阴性对照,代表最高 浓度测试化合物浓度的最终dmso含量)和100μm毛地黄皂苷(阳 性对照,数据标准化设置为0%细胞活力)平行处理,两者均在标准 生长培养基中。
[0079]
48小时孵育结束后,将细胞在标准生长条件下与50μm刃天青 (2.5mm aquabidest.stock)在细胞系特异性生长培养基中孵育4小 时。
[0080]
通过使用spectramax m5多孔板读数器(molecular devices,sanjose,ca,usa)测量了有活力的代谢活性细胞将刃天青转化为试卤 灵(λexc.:540/λem.:590nm)。以生物学一式三份测定数据,每 份都具有技术四份一式。使用graphpad prism 8.0.2版和microsoftexcel 2013进行数据分析。
[0081]
结果与讨论
[0082]
结构解析
[0083]
化合物a(1):黄色固体,hresims:m/z 519.2188[m+h]
+
(计 算值:c
34h31o5+
,519.2166)。1h(400mhz,cdcl3)和
13
c-nmr (100mhz,cdcl3)结果见表1。红外光谱结果为ir(kbr)v
max
: 3310、1655、1610、1420、1000cm-1
,红外光谱结果表明,存在羟基 (3310cm-1
)、芳
香环核心单元(1615cm-1
)和共轭酮(1655cm-1
)。 此外,对1h-nmr谱(表1)解析结果表明,在δ7.33(1h,j=16.0 hz)和7.76(1h,j=16.0hz)处存在信号,这些16.0hz的大耦合 常数是典型的对于反式查耳酮(分别为h-α和h-β)。通过δ127.9 (c-α)和144.1(c-β)处的
13
c-nmr信号以及δ191.8处的反式α、 β-不饱和酮信号进一步证明了查耳酮骨架存在。
[0084]
此外,1h-nmr谱表明,在δ5.59(j=10.0hz,h-2”)、6.76(j =10.0hz,h-1”)和两个磁性等效的δ1.47 6h处的甲基单线态鉴定了 吡喃环的其余部分。在1h-nmr光谱中观察到ab自旋系统,其中δ 6.38和7.47(j=9.0hz)处的两个正交耦合双峰占环a的所有六个 位置。这两个质子的hmbc相关性(图1)已建立分别在环a的h-5' 和h-6'处分配。另一方面,1h-nmr光谱在δ13.74处也具有强氢键 合的低场1-质子信号,并且该羟基在c-2'的位置与其hmbc谱图 c-1'、c-2'和c-3'相关。
[0085]1h-nmr谱还说明δ6.95(d,j=8.0hz,h-5)、7.18(d,j=2.0hz, h-2)和7.2(dd,j=2.0,8.0)处的abx自旋系统。δ6.95、7.18和 7.20处质子的位置被确定为附着在基于环b的hmbc相关性c-5、 c-2和c-6处,δ6.95到c-1、c-3、c-4和c-6,δ7.18至c-1、c-3、 c-4和c-6,δ7.20至c-1、c-2、c-4和c-5。此外,吡喃环在环a 的c-3'和c-4'的位置是通过h-2”到c-3'的hmbc相关性建立。oh-2' 到c-3',h-1”到c-2'、c-3'和c-4'。1h-nmr光谱数据与3,4-二羟基磷 酰胆碱相似,除了δ5.22(4h,s,ch2ph)和7.33-7.49(10h,m,ph) 的两个苄基部分的特定1h-nmr信号。此外,这两个额外的苄基的存 在是根据δ127.1(c-2”'和c-6”')、128.2(c-3”'和c-5”)的
13
c-nmr信 号,以及127.9(c-4”')、136.9(c-1”'),δ70.9和71.5(ch2ph)处 的信号。此外,基于hmbc相关性,将两个苄基置于环a的c-3和 c-4处:δ5.22(4h,ch2ph;两个苄基重叠)至c-3和c-4。基于 光谱结果,化合物1的结构确定为 (e)-3-(3,4-bis(benzyloxy)phenyl)-1-(5-hydroxy-2,2-dimethyl-2h-chrome n-6-yl)prop-2-en-1-one。
[0086]
化合物b(2):黄色固体,hresims:m/z 611.2819[m+h]
+
(计 算值:c
41h39o5+
,611.2792)。红外光谱结果为ir(kbr)v
max
:1605、 1415、1010cm-1
。1h(400mhz,cdcl3)和
13
c-nmr(100mhz, cdcl3)结果见表1。1h-nmr结果表明(表1)具有两个独立的ab 自旋系统,一个在δ6.62(1h,d,j=9.0hz)、7.66(1h,d,j=9.0 hz),另一个在δ5.63(1h,d,j=10.0hz)与6.54(1h,d,j=10.0hz) 耦合。此外,1h-nmr谱表明abx系统在δ6.64(dd,j=2.0,8.0hz)、 6.79(t,j=8.0hz)和7.76(d,j=2.0hz)。从1h-nmr信号中得到 证实,该信号说明了δ5.09(2h,s,ch2ph)、5.05(2h,s,ch2ph)、 4.81(2h,s,ch2ph)和7.30-7.41(15h,m,ph)。通过cozy和hmbc 光谱分析建立了化合物2的完整结构(图1)。基于1d和2d-nmr 研究,化合物b(2)的结构确定为 1-(5-(benzyloxy)-2,2-dimethyl-2h-chromen-6-yl)-3-(3,4-bis(benzyloxy)p henyl)propan-1-one。
[0087]
表1 化合物1和化合物2的1h-nmr(400mhz)和
13
c-nmr(100mhz)数据
1”到c-2'、c-3'和c-4',h-2”到c-3'。此外,化合 物3的nmr数据类似于4-hydroxylonchocarpin,除了异戊二烯基在 δ4.35(2h,d,j=8.0hz)、5.41(1h,m)、1.48(3h,s)和1.65(3h, s)。异戊二烯基在δ73.0(c-1”')、119.4(c-2”')、139.7(c-3”')、 25.7(c-4”')、17.8(c-5”')的
13
c-nmr信号中得到进一步证实。 此外,基于hmbc与信号在δ4.35(h-1”')到c-2'的相关性,o-异 戊二烯基团被置于c-2'。因此,化合物3被鉴定为 (e)-1-(2,2-dimethyl-5-((3-methylbut-2-en-1-yl)oxy)-2h-chromen-6-yl)-3
ꢀ‑
(4-methoxyphenyl)prop-2-en-1-one。
[0092]
表2 化合物3和化合物4的1h-nmr(400mhz)和
13
c-nmr(100mhz)结果
[0093][0094]
化合物d(4):白色固体,hresims:m/z 405.2081[m+h]
+
(计 算值:c
26h29o4+
、405.2060)。红外光谱结果为ir(kbr)v
max
:1600、 1420、1000cm-1
。1h(400mhz,cdcl3)和
13
c-nmr(100mhz, cdcl3)结果见表2。1h-nmr光谱(表2)结果表明,反式查耳酮骨 架在δ7.45(h-α)和7.86(h-β)以及预期的大耦合常数(j=16.0hz)。 此外,
13
c-nmr谱图表明,δ190.6处一个低场信号为典型共轭羰基 碳,证实了查耳酮骨架存在。nmr数据类似于4-hydroxylonchocarpin, 异戊二烯基通过c-异戊二烯化连接到c-5',通过h-1”'在δ3.25
(2h, d,j=8.0hz)处的高场化学位移及其与c-4'、c-5'和c-6'的hmbc 耦合得到证实。此外,存在δ13.65(oh-2')处oh基团的螯合信号, 而没有h-5'信号,δ7.52处的h-6'单线态进一步建立了4环a的结构。 化合物4被鉴定为 (e)-1-(5-hydroxy-2,2-dimethyl-8-(3-methylbut-2-en-1-yl)-2h-chromen-6
ꢀ‑
yl)-3-(4-methoxyphenyl)prop-2-en-1-one。
[0095]
化合物e(5):白色固体,hresims:m/z 473.2706[m+h]
+
(计 算值:c
31h37o4+
、473.2686)。红外光谱结果为ir(kbr)v
max
:1600、 1420、1000cm-1
。1h(400mhz,cdcl3)和
13
c-nmr(100mhz, cdcl3)结果见表3。1h-nmr光谱结果表明(表3),在δ7.43(h-α) 和7.85(h-β)处的反式查耳酮典型双峰信号,具有大耦合常数(j= 16.0hz)。
13
c-nmr谱图表明,δ190.6处的低场信号为典型的共轭 羰基,是查耳酮支架存在的典型特征。nmr数据类似于 4-hydroxylonchocarpin,不同之处在于c-5
′
处的异戊二烯基已通过 c-gernanylation被香叶基取代。以下香叶基1h-nmr光谱信号的存在 证实了这一点:δ3.26(2h,d,j=8.0hz,h-1”')、5.15(1h,m,h-2”')、 1.75(3h,s,h-4”')、2.05(2h,m,h-5”')、2.13(2h,m,h-6”')、5.26 (1h,m,h-7”')、1.59(3h,s,h-9”')和1.66(3h,s,h-10”'),由表 3中
13
c-nmr光谱信号进一步证实。因此,化合物5被确定为 (e)-1-(8-((e)-3,7-dimethylocta-2,6-dien-1-yl)-5-hydroxy-2,2-dimethyl-2 h-chromen-6-yl)-3-(4-methoxyphenyl)prop-2-en-1-one。
[0096]
表3 化合物e的1h-nmr(400mhz)和
13
c-nmr(100mhz)数据
[0097][0098]
细胞毒性评价
[0099]
将分离得到的查尔酮1-6对pc-3(前列腺癌)、mcf-7(乳腺癌)、 mda-mb-468(三阴性乳腺癌细胞系)、sk-n-mc(尤文氏肉瘤)和 hel(红白血病细胞)五种癌细胞的细胞毒性作用测试。所有化合物 使用四种固定浓度进行测试,即10nm、10μm、50μm,最高浓度 为100μm。初步筛选结果显示,在所有测试细胞系中,化合物1-5 在浓度高达100μm时没有显着的抗增殖作用,化合物6除外,在50 μm浓度下可观察到抗增殖活性。
[0100]
因此,进一步研究了化合物6以确定所有五种癌细胞系的ic
50
值。如图3所示,化合物6对sk-n-mc(尤文氏肉瘤)细胞产生细 胞毒作用,ic
50
=25.9μm。针对其他四种癌细胞系hel、mcf-7、 mda-mb-468以及pc-3,化合物6具有细胞抑制/抗增殖活性,ic
50
值为43.5μm、92.2μm、50.4μm和139.2μm。值得注意的是,检 测到的化合物6的抗癌活性在pc-3细胞中最低,并且该细胞系对多 种抗癌剂具有更高的敏感性。然而,化合物6对mda-mb-468(三 阴性乳腺癌细胞系)和sk-n-mc(尤文氏肉瘤)显示出更好的细胞 毒性作用(ic
50
),这些数据对孤儿尤文氏肉瘤和三阴性乳腺癌的治疗 具有潜在的应用前景。
[0101]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明, 对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本 发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应 包含在本发明的保护范围之内。