制备2-(苯基亚氨基)-1,3-噻唑烷-4-酮的方法
1.本发明涉及一种制备通式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮的方法。
2.2-(苯基亚氨基)-1,3-噻唑烷-4-酮和相应的衍生物在制药工业和农业化学工业中作为制备例如手性亚砜的中间体是非常重要的。这种亚砜例如在作物保护中用作杀螨剂(参见例如wo2013/092350或wo2015/150348)。
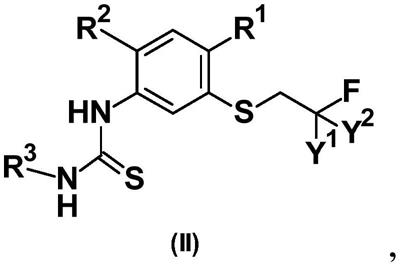
3.2-(苯基亚氨基)-1,3-噻唑烷-4-酮的化学合成是已知的。例如,这可通过使通式(ii)的适当取代的硫脲与通式(iii)的乙酸衍生物反应来实现(参见例如wo2013/092350;ep 985670;advances in heterocycl.chem.25,(1979)85)。原则上有多种制备通式(ii)的硫脲的方法。一种简单而有效的方法包括使通式(iv)的适当取代的苯胺与通式(v)的异硫氰酸酯反应(wo2014/202510)。相反,也可通过使通式(vi)的芳基异硫氰酸酯与通式(vii)的苯胺反应来获得通式(ii)的硫脲(jp2011/042611)。
4.因此,制备通式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮的常见方法的特征在于,在第一步中,使通式(iv)的苯胺与通式(v)的异硫氰酸酯反应,或使通式(vi)的芳基异硫氰酸酯与通式(vii)的胺反应,然后例如通过过滤分离由此形成的通式(ii)的硫脲。在已知方法的第二步中,然后使通式(ii)的硫脲与通式(iii)的乙酸衍生物在碱的存在下反应以形成通式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮。
5.该方法的缺点是涉及分离硫脲中间体的两个单独步骤的费力过程。这是耗时的,并且导致高成本。此外,取决于所使用的稀释剂的性质,这可能导致通式(ii)的硫脲的沉淀物,所述沉淀物的体积可能太大,以至于反应混合物变得无法搅拌,并且不能从反应容器中排出。如果发生这种情况,则硫脲中间体的分离实际上变得不可能。此外,当受到热应力时,例如也可能发生在过滤后干燥固体时,已知硫脲(synthesis 1984,825-7;wo2014/189753;j.labelled comp.and radiopharmaceuticals 22(1985)313-27)经历部分热裂解至起始化合物(热不稳定性)。
6.现有技术中已知的方法(a)示于方案(1)中,其中x、y1、y2、w、r1、r2和r3如下文所定义。
7.方案(1)
[0008][0009]
鉴于上述缺点,迫切需要一种简化的、工业上和经济上可行的制备通式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮的方法。可使用这种方法获得的2-(苯基亚氨基)-1,3-噻
唑烷-4-酮应优选以高收率和高纯度提供。特别地,所寻求的方法应当允许获得所需的目标化合物,而无需复杂的分离方法。此外,所寻求的方法应当明显缩短反应时间,并优选允许使用适于工业规模使用的稀释剂。
[0010]
出人意料地,发现通式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮可通过使通式(iv)的苯胺与通式(v)的异硫氰酸酯在通式(iii)的乙酸衍生物和碱的存在下反应来制备,其中作为中间体形成的通式(ii)的硫脲直接且优选原位反应以形成2-(苯基亚氨基)-1,3-噻唑烷-4-酮。
[0011]
因此,本发明提供一种制备通式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮的方法,
[0012][0013]
其中
[0014]
y1和y2独立地为氟、氯或氢,
[0015]
r1和r2独立地为氢、c
1-c
12
烷基、c
1-c
12
卤代烷基、氰基、卤素或硝基,和
[0016]
r3为任选取代的c
6-c
10
芳基、c
1-c
12
烷基或c
1-c
12
卤代烷基,其中取代基选自卤素、c
1-c6烷基、c
3-c
10
环烷基、氰基、硝基、羟基、c
1-c6烷氧基、c
1-c6卤代烷基和c
1-c6卤代烷氧基,特别地选自氟、氯、c
1-c3烷基、c
3-c6环烷基、环丙基、氰基、c
1-c3烷氧基、c
1-c3卤代烷基和c
1-c3卤代烷氧基,
[0017]
其特征在于,使式(iv)的苯胺
[0018][0019]
其中y1、y2、r1和r2如上文所定义,
[0020]
在式(iii)的乙酸衍生物的存在下
[0021][0022]
其中
[0023]
x为溴、氯、oso2me、oso2ph、oso2(4-me-ph)或oso2cf3,和
[0024]
w为oh或o(c
1-c6烷基)基团,
[0025]
以及在碱的存在下,首先与式(v)的异硫氰酸酯反应
[0026][0027]
其中
[0028]
r3如上文所定义,
[0029]
以形成式(ii)的硫脲
[0030][0031]
其中y1、y2、r1、r2和r3如上文所定义,
[0032]
然后将式(ii)的硫脲转化成式(i)的化合物,其中在向反应混合物中加入至少一种式(iv)和(v)的化合物之前,式(iii)的乙酸衍生物最初存在于反应混合物中。
[0033]
因此,当式(iv)的苯胺与式(v)的异硫氰酸酯反应以形成式(ii)的硫脲时,式(iii)的乙酸衍生物已经存在。这对该反应无不良影响;相反,其确保——而非在反应混合物中累积——式(ii)的硫脲立即进一步转化成式(i)的化合物。换句话说,式(ii)的硫脲立即原位转化成式(i)的化合物,即作为中间体形成的式(ii)的硫脲立即进一步原位反应以形成式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮。
[0034]
式(i)的化合物可以作为e-或z-异构体或作为这些异构体的混合物存在。这由式(i)中的交叉双键表示。在本发明的一个单独的实施方案中,所述化合物在每种情况下为e-异构体的形式。在本发明的另一个单独的实施方案中,所述化合物在每种情况下为z-异构体的形式。在本发明的另一个单独的实施方案中,化合物为e-和z-异构体的混合物形式。在本发明的一个优选的单独实施方案中,化合物为z-异构体的形式或e-和z-异构体的混合物的形式,其中z-异构体的比例大于50%,并且渐增地优选大于60%、65%、70%、75%、80%、85%、90%、95%,基于混合物中e-和z-异构体的总量计。
[0035]
在上述式(i)、(ii)、(iii)、(iv)和(v)中列出的基团x、y1、y2、w、r1、r2和r3的优选的、特别优选的和非常特别优选的定义说明如下。
[0036]
优选:
[0037]
x为溴或氯,
[0038]
y1和y2独立地为氟、氯或氢,
[0039]
w为o(c
1-c6烷基)基团,
[0040]
r1和r2独立地为氟、氯、c
1-c3烷基或氢,和
[0041]
r3为任选取代的苯基、c
1-c6烷基或c
1-c6卤代烷基,其中取代基选自卤素、c
1-c6烷基、c
3-c
10
环烷基、氰基、硝基、羟基、c
1-c6烷氧基、c
1-c6卤代烷基和c
1-c6卤代烷氧基,特别地选自氟、氯、c
1-c3烷基、c
3-c6环烷基、环丙基、氰基、c
1-c3烷氧基、c
1-c3卤代烷基和c
1-c3卤代烷氧基。
[0042]
特别优选:
[0043]
x为溴或氯,
[0044]
y1和y2独立地为氟或氢,
[0045]
w为o(c
1-c6烷基)基团,
[0046]
r1和r2独立地为氟、氯、氢或甲基,和
[0047]
r3为c
1-c6烷基或c
1-c6卤代烷基。
[0048]
非常特别优选:
[0049]
x为溴或氯,
[0050]
y1和y2为氟,
[0051]
w为och3或oc2h5基团,
[0052]
r1和r2独立地为氟、氢或甲基,和
[0053]
r3为c
1-c6卤代烷基。
[0054]
最优选:
[0055]
x为溴或氯,
[0056]
y1和y2为氟,
[0057]
w为och3,
[0058]
r1为甲基,
[0059]
r2为氟,和
[0060]
r3为ch2cf3。
[0061]
出人意料地,式(i)的2-(苯基亚氨基)-1,3-噻唑烷-4-酮可以通过本发明的方法以良好的收率和高纯度制备。本发明的方法允许式(iv)的苯胺与式(v)的异氰酸酯在碱和式(iii)的乙酸衍生物的存在下以高选择性和高收率进行反应的事实是出人意料的,因为已知苯胺与式(iii)的乙酸衍生物在氮上发生烷基化(参见例如us20050020645;wo2004/039764)。在本发明的方法中,这种出乎意料的结果没有发生至任何可察觉的程度;相反,在形成式(ii)的硫脲时已经存在的式(iii)的乙酸衍生物导致式(ii)的硫脲立即进一步转化成式(i)的化合物。这避免了形成难以处理的粘性、糊状反应混合物。无法预见式(iii)的乙酸衍生物对化合物(iv)和(v)的形成式(ii)的化合物的反应几乎没有或没有影响,从而可以在早期阶段加入到反应混合物中,并因此可立即用于式(ii)的硫脲的反应。因此,这带来了式(i)的目标化合物的纯度和收率的改善,并且重要的是,工艺经济性的改善,特别是在工业规模上。此外,本发明的方法允许使用适用于工业规模生产的稀释剂,特别是其中可能出现大量式(ii)的硫脲沉淀物的稀释剂。本发明方法带来的工艺经济性的另一个优点是它允许获得所需的目标化合物而无需复杂的中间体分离程序。
[0062]
本发明的方法可以基于以下方案(2)来阐明,其中x、y1、y2、w、r1、r2和r3如上文所定义。方案(2)说明了清楚的转化。如所述,在向反应混合物加入至少一种式(iv)和(v)的化合物之前,式(iii)的化合物存在于反应混合物中。
[0063]
方案(2)
[0064][0065]
一般定义
[0066]
在本发明的上下文中,除非另有定义,否则术语“卤素(hal)”涵盖选自氟、氯、溴和碘的元素,优选使用氟、氯和溴,并且特别优选使用氟和氯。
[0067]
任选取代的基团可以被单取代或多取代;如果被多取代,则取代基可相同或不同。除非在相关位置另有说明,否则取代基选自卤素、c
1-c6烷基、c
3-c
10
环烷基、氰基、硝基、羟基、c
1-c6烷氧基、c
1-c6卤代烷基和c
1-c6卤代烷氧基,特别地选自氟、氯、c
1-c3烷基、c
3-c6环烷基、环丙基、氰基、c
1-c3烷氧基、c
1-c3卤代烷基和c
1-c3卤代烷氧基。
[0068]
被一个或多个卤素原子(hal)取代的烷基例如选自三氟甲基(cf3)、二氟甲基(chf2)、cf3ch2、clch2或cf3ccl2。
[0069]
除非另有不同定义,否则在本发明上下文中的烷基为直链、支链或环状饱和烃基。
[0070]
定义c
1-c
12
烷基涵盖本文中对烷基限定的最宽范围。具体地,该定义涵盖例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基和叔丁基、正戊基、正己基、1,3-二甲基丁基、3,3-二甲基丁基、正庚基、正壬基、正癸基、正十一烷基、正十二烷基。
[0071]
除非另有不同定义,否则在本发明上下文中的芳基为芳族烃基,其可包括0个、1个、2个或更多个杂原子(选自o、n、p和s)。
[0072]
具体地,该定义涵盖例如环戊二烯基、苯基、环庚三烯基、环辛四烯基、萘基和蒽基;2-呋喃基、3-呋喃基、2-噻吩基、3-噻吩基、2-吡咯基、3-吡咯基、3-异噁唑基、4-异噁唑基、5-异噁唑基、3-异噻唑基、4-异噻唑基、5-异噻唑基、3-吡唑基、4-吡唑基、5-吡唑基、2-噁唑基、4-噁唑基、5-噁唑基、2-噻唑基、4-噻唑基、5-噻唑基、2-咪唑基、4-咪唑基、1,2,4-噁二唑-3-基、1,2,4-噁二唑-5-基、1,2,4-噻二唑-3-基、1,2,4-噻二唑-5-基、1,2,4-三唑-3-基、1,3,4-噁二唑-2-基、1,3,4-噻二唑-2-基和1,3,4-三唑-2-基、1-吡咯基、1-吡唑基、1,2,4-三唑-1-基、1-咪唑基、1,2,3-三唑-1-基、1,3,4-三唑-1-基、3-哒嗪基、4-哒嗪基、2-嘧啶基、4-嘧啶基、5-嘧啶基、2-吡嗪基、1,3,5-三嗪-2-基和1,2,4-三嗪-3-基。
[0073]
式(iv)的苯胺向式(i)的化合物的转化优选在稀释剂的存在下进行。本发明方法中的合适的稀释剂特别是以下物质:四氢呋喃(thf)、二噁烷、乙醚、甲基叔丁基醚(mtbe)、叔戊基甲基醚(tame)、2-甲基-thf、乙腈(acn)、丙酮、丁腈、乙酸乙酯、乙酸异丙酯、乙酸丁酯、乙酸戊酯、甲基异丁基酮、碳酸亚乙酯、碳酸亚丙酯、n,n-二甲基乙酰胺(dmac)、n,n-二甲基甲酰胺(dmf)、n-甲基吡咯烷酮、二甲亚砜(dmso)、环丁砜、四氯乙烯、四氯乙烷、二氯丙烷、二氯甲烷(二氯甲烷、dcm)、二氯丁烷、氯仿、四氯化碳、三氯乙烷、三氯乙烯、五氯乙烷、
1,2-二氯乙烷、甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯、均三甲苯、氯苯、1,2-二氯苯、苯甲醚、正戊烷、正己烷、正庚烷、正辛烷、1,2,4-三甲基戊烷(异辛烷)、石油醚40/55、特殊沸点油精(special boiling point spirit)80/110、环己烷或甲基环己烷。也可以使用所述稀释剂的混合物。
[0074]
本发明方法中优选的稀释剂为二氯甲烷、氯仿、1,2-二氯乙烷、乙腈、丙酮、乙酸乙酯、甲基叔丁基醚(mtbe)、四氢呋喃(thf)、2-甲基-thf、n,n-二甲基乙酰胺(dmac)、n,n-二甲基甲酰胺(dmf)、甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯、均三甲苯、氯苯、1,2-二氯苯、苯甲醚、正庚烷、正辛烷、1,2,4-三甲基戊烷(异辛烷)、石油醚40/55、特殊沸点油精80/110、甲基环己烷或所述稀释剂的混合物。
[0075]
特别优选的稀释剂为乙腈、乙酸乙酯、四氢呋喃(thf)、甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯、均三甲苯、氯苯、1,2-二氯苯、苯甲醚、正庚烷、1,2,4-三甲基戊烷(异辛烷)、石油醚40/55、特殊沸点油精80/110、甲基环己烷或所述稀释剂的混合物。非常特别优选甲苯、邻二甲苯、间二甲苯、对二甲苯、乙苯或氯苯或所述稀释剂的混合物。
[0076]
式(v)的异硫氰酸酯优选以0.95:1至2:1的摩尔比使用,基于式(iv)的苯胺计。进一步优选1.01:1至1.5:1的摩尔比,同样在每种情况下基于式(iv)的苯胺计。
[0077]
本发明方法中使用的碱可以是有机碱或无机碱。有机碱的实例为三甲胺、三乙胺、三丁胺和乙基二异丙胺。无机碱的实例为乙酸钾、乙酸钠、氢氧化锂、氢氧化钾、氢氧化钠、碳酸氢钾、碳酸氢钠、碳酸钾、碳酸钠、碳酸铯、碳酸钙和碳酸镁。优选氢氧化钾、氢氧化钠、碳酸钾和碳酸钠。特别优选碳酸钾。
[0078]
在本发明的方法中,碱优选以0.8:1至3:1的摩尔比使用,基于式(iv)的苯胺计。进一步优选1:1至2:1的摩尔比,同样在每种情况下基于式(iv)的苯胺计。
[0079]
在本发明的方法中,式(iii)的乙酸衍生物优选以0.9:1至2:1的摩尔比使用,基于式(iv)的苯胺计。进一步优选1.0:1至1.5:1的摩尔比,同样在每种情况下基于式(iv)的苯胺计。
[0080]
本发明的方法通常在-20℃至150℃、优选0℃至120℃、最优选5℃至80℃的温度下进行。
[0081]
反应通常在标准压力下进行,但也可以在加压或减压下进行。
[0082]
所需的式(i)的化合物可以例如通过随后的过滤或萃取来分离。这种方法是本领域技术人员已知的。
[0083]
通过随后的实施例详细说明本发明,但不应以它们限制本发明的方式来解释这些实施例。
实施例:
[0084]
实施例1:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟甲基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮在甲苯中的合成
[0085]
将648.8g甲苯、153.9g[1.09mol]的1,1,1-三氟-2-异硫氰酸根合乙烷、170.3g[1.23mol]碳酸钾和165.9g[1.09mol]溴乙酸甲酯装入反应容器中。伴随搅拌将反应混合物加热至50℃。在此温度下,在30分钟内滴加235.8g[0.986mol]2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺在235.8g甲苯中的溶液,同时继续搅拌。然后将反应混合物在50℃下
搅拌7小时,在2小时内冷却至20℃,并在20℃下另搅拌12小时。在这段时间内,反应混合物是易于搅拌的悬浮液。对于后处理,伴随搅拌将反应混合物计量加入到672.8g水中。其后用259.5g甲苯冲洗反应容器,并将冲洗液同样计量加入到水中。分离出上层有机相,并与270g盐酸(16%)一起搅拌。重新进行的相分离得到1523.3g有机相,相对于参考标准的定量hplc分析显示,其含有26.0%(w/w)的目标化合物(396.1g,相当于理论值95.6%的收率)。
[0086]
实施例2:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮在甲基环己烷中的合成
[0087]
将100ml甲基环己烷(mch)、7.76g[55mmol]1,1,1-三氟-2-异硫氰酸根合乙烷、8.41g[55mmol]溴乙酸甲酯和8.6g[62.5mmol]碳酸钾加入到反应容器中。将混合物加热至50℃,并在此温度下伴随搅拌滴加11.9g[50mmol]2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺,在50℃下继续搅拌24小时。反应期间粘性固体的少量沉积不会不利地影响反应混合物的可搅拌性。在这段时间结束时,存在微红色的、易于搅拌的悬浮液。将其冷却至室温,然后与100ml的1n盐酸一起搅拌,然后分离相并浓缩有机相。这提供了10g产物,其hplc纯度为80.3%,相当于理论值38.2%的收率。然后用三份100ml的mch萃取水相。浓缩合并的有机相。这提供了9.8g产物,其hplc纯度为71.8%,相当于理论值33.5%的收率。因此总收率为理论值的71.7%。
[0088]
实施例3:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟甲基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮在二甲苯中的合成
[0089]
将17.2g工业二甲苯混合物和5.18g[37.5mmol,1.5当量]碳酸钾装入到反应容器中。加入4.21g[27.5mmol,1.1当量]溴乙酸甲酯,然后用2.15g二甲苯冲洗。滴加3.91g[27.5mmol,1.1当量]的1,1,1-三氟-2-异硫氰酸根合乙烷,然后用2.15g二甲苯冲洗。伴随搅拌将反应混合物加热至50℃。在该温度下,伴随搅拌在30分钟内滴加6.16g[25.0mmol,1.0当量]的2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺。然后将反应混合物在50℃下搅拌6.5小时,通过hplc定期检查转化率。在这段时间内,反应混合物是易于搅拌的悬浮液。对于后处理,将反应混合物冷却至室温并加入15g水。将混合物转移到分液漏斗中,然后用3ml二甲苯冲洗。相分离得到35.1g深棕色二甲苯溶液,相对于参考标准的定量hplc分析显示,其含有29.0%(w/w)的标题化合物(10.18g,相当于理论值96.9%的收率)。
[0090]
实施例4:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮在氯苯中的合成
[0091]
将22.1g氯苯和5.18g[37.5mmol,1.5当量]碳酸钾装入到反应容器中。加入4.21g[27.5mmol,1.1当量]溴乙酸甲酯,然后用2.15g氯苯冲洗。滴加3.91g[27.5mmol,1.1当量]的1,1,1-三氟-2-异硫氰酸根合乙烷,然后用2.8g氯苯冲洗。伴随搅拌将反应混合物加热至50℃。在该温度下,伴随搅拌在30分钟内滴加6.16g[25.0mmol,1.0当量]的2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺。然后将反应混合物在50℃下搅拌6.5小时,通过hplc定期检查转化率。在这段时间内,反应混合物是易于搅拌的悬浮液。对于后处理,将反应混合物冷却至室温并加入15g水。将混合物转移到分液漏斗中,然后用3ml氯苯冲洗。相分离得到42.1g深棕色氯苯溶液,相对于参考标准的定量hplc分析显示,其含有23.5%(w/w)的标题化合物(9.89g,相当于理论值94.1%的收率)。
[0092]
比较实施例:
[0093]
比较实施例1:1-{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}-3-(2,2,2-三氟乙基)硫脲在甲苯中的合成
[0094]
将5.0g 2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺[20.9mmol,1.0当量]加入到30ml甲苯中,并在室温下向其中滴加3.2g 1,1,1-三氟-2-异硫氰酸根合乙烷[23.0mmol,1.1当量]。将反应混合物在室温下搅拌3小时,导致原始溶液形成非常稠的、难以搅拌的悬浮液。监测反应表明仅约85%转化率。将反应混合物加热至50℃以使其可再次部分搅拌。在50℃下3小时后,仍未实现完全转化,因此将反应混合物加热至70℃。即使在70℃下3小时后仍未实现完全转化(hplc监测反应表明仍存在0.9%的苯胺)。将反应混合物冷却至5℃,并将非常稠的糊状悬浮液尽可能彻底地转移至吸滤器中,并分离固体。将获得的固体用冷mtbe洗涤并在减压下干燥。得到5.1g目标产物,为米色固体(理论值的61%)。浓缩滤液,另外得到2.2g棕色固体,其目标产物含量为约60%(理论值的17%)。差的分离收率部分还归因于在将非常稠的悬浮液转移至吸滤器期间的相对大的损失。
[0095]
比较实施例2:1-{2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}-3-(2,2,2-三氟乙基)硫脲在甲基环己烷中的合成
[0096]
将77ml甲基环己烷(mch)和11.9g[50mmol]2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺装入到反应容器中。将其加热至50℃,并在该温度下伴随搅拌在大约5分钟内滴加8.1g[57.5mmol]1,1,1-三氟-2-异硫氰酸根合乙烷。在几分钟后,目标产物开始沉淀出来,导致反应混合物变成稠的、不可搅拌的糊状物。即使再加入80ml甲基环己烷也不能使混合物再次可搅拌。将反应混合物冷却至20℃,并用大量mch从反应容器中冲洗出来。抽滤出固体,用mch洗涤并干燥。这提供了18.55g产物,hplc分析纯度为98.5%(a/a),相当于理论值96%的收率。因此,尽管收率非常好,但反应混合物的极其糊状的稠度使得该方法在工业规模上不可行。
[0097]
比较实施例3:(2z)-2-({2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯基}亚氨基)-3-(2,2,2-三氟乙基)-1,3-噻唑烷-4-酮在甲苯中的合成
[0098]
将7.1g 1,1,1-三氟-2-异硫氰酸根合乙烷[95%,48.0mmol,1.2当量]溶解在40ml甲苯中,并与9.57g 2-氟-4-甲基-5-[(2,2,2-三氟乙基)硫烷基]苯胺(40.0mmol,1.1当量)一起在20℃下搅拌(400rpm)30分钟,导致淡黄色溶液形成含有白色固体的悬浮液。在1小时后,悬浮液不再可搅拌,但通过悬浮液的hplc分析监测反应表明仅约65%的转化率。再加入10ml甲苯,将搅拌速度增加至600rpm,并将反应混合物加热至40℃,其结果是混合物再次变得可适度搅拌。在40℃下3小时后(反应的hplc监测显示约87%转化率),加入8.3g固体碳酸钾[60.0mmol,1.5当量]。再过30分钟后,在40℃下在1小时内加入8.0g[52.0mmol,1.3当量]2-溴乙酸甲酯,并将反应混合物在40℃下搅拌20小时,导致形成溴化钾和碳酸钾在目标产物的甲苯溶液中的悬浮液,其再次可容易地搅拌。此时对反应的hplc监测显示苯胺完全转化,并且仅有痕量中间体硫脲。将反应混合物冷却至20℃,在20℃下再搅拌17小时并过滤。用少量甲苯洗涤固体,并将合并的滤液浓缩至66.8g红棕色甲苯溶液,相对于外部标准的hplc显示,其含有21.1%的目标产物(理论值的84%),并且既不含苯胺也不含硫脲中间体。