1.本发明涉及光刻技术领域,具体涉及一种硬掩膜组合物及其制备方法、形成图案的方法。
背景技术:
2.近年来,半导体工业己开发具有数纳米至数十纳米尺寸图案的超精细技术。为实现如此精细化的图案,一方面光刻用光源的波长向短波化方向发展,例如由先前广泛使用的水银灯的g-line(436nm)、i-line(365nm),向krf受激准分子激光器(248nm)、arf受激准分子激光器(193nm)发展;另一方面,为了防止精细化的光致抗蚀剂图案溃散,光致抗蚀剂膜的厚度在逐渐减薄。相应地,也产生两方面的问题,一是伴随着曝光波长的短波长化,焦点深度减小,二是减薄的光致抗蚀剂图案难以提供足够耐刻蚀性来刻蚀材料层。针对第一方面的问题,为了高精度地形成所期望的抗蚀剂图案,提高在基板上形成的被膜的平坦化性变得很重要,即能在基板上形成无高低差的平坦的涂膜的抗蚀剂下层膜是必不可少的;针对第二方面的问题,需要在光致抗蚀剂与材料层之间引入耐刻蚀强的无机物膜或有机物膜,即抗蚀剂下层膜或硬掩膜。作为硬掩膜的无机物通常由氮化硅、氮氧化硅、多晶硅、氮化硅、无定型碳等,通常需要通过化学气相沉积(cvd)法制备,具有耐刻蚀性优良的特点,但同时也存在颗粒问题和设备费用投入高昂等问题。为解决这些问题,提出了使用旋涂的有机物硬掩膜替代上述化学气相沉积的无机硬掩膜的方法。
3.上述有机物硬掩膜的典型应用工艺为:在材料层上旋涂高碳含量的有机硬掩膜层;之后在其上形成硅层,该硅层可以通过旋涂或化学气相沉积形成;最后在硅层上旋涂光致抗蚀剂层。硅在使用氧气刻蚀时具有高度的耐刻蚀性,并且通过在硅层下方提供高碳含量的有机掩膜层,可以获得非常大的长径比。因此,该高碳含量有机硬掩膜层比其上方的硅层和光致抗蚀剂层厚得多。
4.高碳含量有机硬掩膜组合物通常由高碳含量树脂、交联剂、催化剂、添加剂以及溶剂组成。其中,高碳含量树脂通常包含大量的芳香基团,一方面可以带来优异的耐刻蚀性能;另一方面,大量的芳香基团导致树脂结构的刚性太强,在常用的有机硬掩膜组合物溶解性变差;同时,树脂的流动性变差,无法均匀涂覆在衬底表面,尤其是已经图案化的衬底表面。
技术实现要素:
5.有鉴于此,本发明提供了一种硬掩膜组合物及其制备方法、形成图案的方法。本发明中提供的硬掩膜组合物可确保溶剂的可溶性、间隙填充特性和平坦化特性并兼具优异耐刻蚀性。
6.为解决上述技术问题,本发明采用以下技术方案:
7.第一方面,本发明提供了一种硬掩膜组合物,所述硬掩膜组合物中包含聚合物和溶剂,所述聚合物为化学式1所示的化合物:
[0008][0009]
其中,r1选自氢原子、碳原子为1-6的烷基、碳原子数为6-30的取代或非取代的芳基中的任意一种;ar1选自碳原子数为6-18的取代或非取代的联苯基团、被至少一个氧原子隔开的为碳原子数为6-18的取代或非取代的联苯基团中的任意一种;n取1-100之间任意整数。
[0010]
进一步地,当所述r1选自碳原子数为6-30的取代的芳基中的任意一种时,所述取代的芳基中取代的基团包括:苯基、含有取代基的苯基、萘基、含有取代基的萘基;
[0011]
当ar1选自碳原子数为6-18的取代或非取代的联苯基团的任意一种时,所述取代的联苯基团中取代的基团包括:甲基、乙基、丙基、丁基、叔丁基;
[0012]
当ar1选自被至少一个氧原子隔开的为碳原子数为6-18的取代或非取代的联苯基团中的任意一种时,所述取代的联苯基团中取代的基团包括:甲基、乙基、丙基、丁基、叔丁基。
[0013]
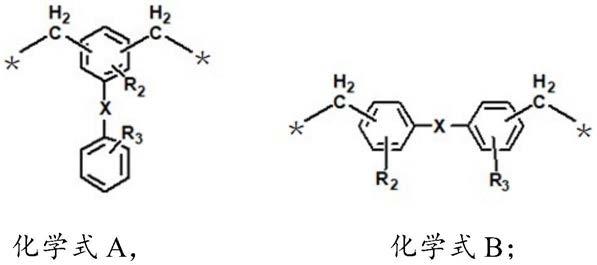
进一步地,所述ar1选自族群1所示二价基团中的任意一种,所述族群1包括化学式a和化学式b所示的化合物:
[0014][0015]
其中,r2、r3各自独立地选自氢、羟基、甲氧基、乙氧基、卤素、碳原子数为1-6的取代或非取代的烷基、碳原子数为6-12的取代或非取代的芳基中的任意一种,x为氧原子或单键,*表示连接点。
[0016]
进一步地,所述溶剂包括丙二醇单甲醚乙酸酯、丙二醇单甲醚、环己酮及乳酸乙酯中的任意一种。
[0017]
进一步地,,所述硬掩膜组合物中还包含交联剂、催化剂、表面活性剂、增塑剂中的至少一种。
[0018]
进一步地,以所述硬掩膜组合物的总量计,所述聚合物的质量占比为4%~25%,所述交联剂的质量占比0.4~3%,所述催化剂的质量占比0.001%~0.05%,所述表面活性剂的质量占比0.01%~0.1%,所述增塑剂的质量占比0%~2.5%,所述溶剂的质量占比75~95.6%。
[0019]
第二方面,本发明提供了如上所述的硬掩膜组合物的制备方法,所述制备方法为:取所述聚合物与溶剂混合,所述聚合物的质量占比为4%~25%,溶剂的质量占比75~
95.6%。
[0020]
进一步地,所述制备方法为:取所述聚合物与所述溶剂、交联剂、催化剂、表面活性剂以及增塑剂混合,以所述硬掩膜组合物的总量计,所述聚合物的质量占比为4%~25%,所述交联剂的质量占比0.4~3%,所述催化剂的质量占比0.001%~0.05%,所述表面活性剂的质量占比0.01%~0.1%,所述增塑剂的质量占比0%~2.5%,所述溶剂的质量占比75~95.6%。
[0021]
第三方面,本发明提供了一种形成图案的方法,所述方法包括以下步骤:
[0022]
在基板上提供材料层;
[0023]
将如上所述的硬掩膜组合物施加在所述材料层上;
[0024]
热处理所述硬掩膜组合物以形成硬掩膜;
[0025]
在所述硬掩膜上形成含硅薄层;
[0026]
在所述含硅薄层上形成光刻胶抗蚀层;
[0027]
将所述光刻胶抗蚀层曝光并显影以形成光刻胶图案;
[0028]
使用所述光刻胶图案选择性去除所述含硅薄层和所述硬掩膜以暴露所述材料层的一部分;
[0029]
刻蚀所述材料层的暴露部分。
[0030]
进一步地,其中形成硬掩膜的方法为:将所述硬掩膜组合物以溶液形式旋涂在所述材料层上,在200℃到500℃下对所述硬掩膜组合物进行热处理,热处理时间约为10秒到10分钟,以形成硬掩膜。
[0031]
本发明的上述技术方案的有益效果如下:
[0032]
本发明提供了一种硬掩膜组合物及其制备方法、形成图案的方法,本发明的硬掩膜组合物中包含一种聚合物,所述聚合物中包含由芘衍生物与具有联苯或醚基的化合物缩合而成。由于联苯和醚基的旋转特性,使得上述聚合物的柔性增加,从而能够提高硬掩膜组合物的溶解性和涂覆性,可以应用于填充在已图案化的材料层中开放的构形特征,例如通孔、沟槽。另外,由于聚合物包含高碳含量的芘衍生物,从而使硬掩膜具有更优的耐刻蚀性。
具体实施方式
[0033]
为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明的限制。
[0034]
第一方面,本发明提供了一种硬掩膜组合物,所述硬掩膜组合物中包含一种聚合物和溶剂,所述聚合物为化学式1所示的化合物:
[0035][0036]
其中,r1选自氢原子、碳原子为1-6的烷基、碳原子数为6-30的取代或非取代的芳基中的任意一种;ar1选自碳原子数为6-18的取代或非取代的联苯基团、被至少一个氧原子
隔开的为碳原子数为6-18的取代或非取代的联苯基团中的任意一种;n取1-100之间任意整数。
[0037]
本发明的硬掩膜组合物中包含一种聚合物,所述聚合物中包含由芘衍生物与具有联苯或醚基的化合物缩合而成。由于联苯和醚基的旋转特性,使得上述聚合物的柔性增加,从而能够提高硬掩膜组合物的溶解性和涂覆性,可以应用于填充在已图案化的材料层中开放的构形特征,例如通孔、沟槽。另外,由于聚合物包含高碳含量的芘衍生物,从而使硬掩膜具有更优的耐刻蚀性。
[0038]
根据本发明的一些实施例,当所述r1选自碳原子数为6-30的取代的芳基中的任意一种时,所述取代的芳基中取代的基团包括:苯基、含有取代基的苯基、萘基、含有取代基的萘基;
[0039]
当ar1选自碳原子数为6-18的取代或非取代的联苯基团的任意一种时,所述取代的联苯基团中取代的基团包括:甲基、乙基、丙基、丁基、叔丁基;
[0040]
当ar1选自被至少一个氧原子隔开的为碳原子数为6-18的取代或非取代的联苯基团中的任意一种时,所述取代的联苯基团中取代的基团包括:甲基、乙基、丙基、丁基、叔丁基。
[0041]
根据本发明的一些实施例,所述ar1选自族群1所示二价基团中的任意一种,所述族群1包括化学式a和化学式b所示的化合物:
[0042][0043]
其中,r2、r3各自独立地选自氢、羟基、甲氧基、乙氧基、卤素、碳原子数为1-6的取代或非取代的烷基、碳原子数为6-12的取代或非取代的芳基中的任意一种,x为氧原子或单键,*表示连接点。
[0044]
具体地,本发明中提供的硬掩膜组合物中包含的一种聚合物,所述聚合物可以通过由化学式2所示的芘衍生物和族群2中所示的二醇衍生物,在磺酸等酸性催化剂的存在下在溶剂中进行聚合反应来合成,其中族群2中所示的二醇衍生物包括如化学式c和化学式d所示的化合物:
[0045][0046]
其中,r1选自氢原子、碳原子为1-6的烷基、碳原子数为6-30的取代或非取代的芳基中的任意一种;r2、r3各自独立地选自氢、羟基、甲氧基、乙氧基、卤素、碳原子数为1-6的取代或非取代的烷基、碳原子数为6-12的取代或非取代的芳基中的任意一种,x为氧原子或单键。
[0047]
作为上述缩聚反应中使用的酸催化剂,可以列举如硫酸、磷酸、高氯酸等无机酸,也可以列举对苯甲磺酸、甲酸、草酸等有机酸。当含有芳香环的聚合物质量数为100份时,酸催化剂的用量为0.1-100质量份。
[0048]
上述缩聚反应的通常在溶剂中进行。作为溶剂,只要不阻碍反应,均可以使用,可列举出四氢呋喃、丙二醇单甲醚、丙二醇二甲醚等。此外,所使用的酸催化剂如果为甲酸那样的液态物质,则其也可以兼做溶剂。
[0049]
进一步地,所述缩聚反应的温度通常为40-200℃,反应时间根据反应温度和分子量要求进行选择,通常为30分钟到50小时左右。
[0050]
根据本发明的一些实施例,所述溶剂包括丙二醇单甲醚乙酸酯、丙二醇单甲醚、环己酮及乳酸乙酯中的任意一种。
[0051]
根据本发明的一些实施例,所述硬掩膜组合物中还包含交联剂、催化剂、表面活性剂、增塑剂中的至少一种。
[0052]
根据本发明的一些实施例,以所述硬掩膜组合物的总量计,所述聚合物的质量占比为4%~25%,所述交联剂的质量占比0.4~3%,所述催化剂的质量占比0.001%~0.05%,所述表面活性剂的质量占比0.01%~0.1%,所述增塑剂的质量占比0%~2.5%,所述溶剂的质量占比75~95.6%。
[0053]
根据本发明中的一些实施例,所述交联剂包括甘脲化合物、环氧化合物、三聚氰胺和三聚氰胺衍生物中的任意一种或两种以上。其中,甘脲化合物可以为如化学式3所示的化合物,环氧化合物可以为如化学式4所示的化合物,三聚氰胺和三聚氰胺衍生物可以为如化学式5所示的化合物:
[0054][0055]
进一步地,所述交联剂可以使用耐热性高的交联剂。作为耐热性高的交联剂,可以优选使用在分子内具有苯环和萘环等的芳香环的化合物,可列举出具有化学式6所示的化合物和化学式7所示的结构单元的聚合物或低聚物。
[0056][0057]
其中,r7选自氢原子、碳原子为1-6的烷基或碳原子数为6-30的取代或非取代的芳基中的任意一种;r8、r9各自独立地选自氢原子、碳原子为1-6的烷基中的任意一种;n1取1~5之间的整数;n2取1~3之间的整数;具有化学式7结构单元的聚合物或低聚物,优选地重复单元为2~50。
[0058]
根据本发明中的一些实施例,所述催化剂一般为酸性化合物,起促进交联反应的作用,所述催化剂包括化学式1-a~化学式1-f中的任意一种或两种以上,所述化学式1-a~化学式1-f如下所示:
[0059][0060]
根据本发明中的一些实施例,所述表面活性剂包括聚氧乙烯烷基醚类、聚氧乙烯烷基芳基醚类、失水山梨糖醇脂肪酸酯类、聚氧乙烯失水山梨糖醇脂肪酸酯类中的任意一种或两种以上。
[0061]
根据本发明中的一些实施例,所述增塑剂主要是为了提高硬掩膜组合物的流动性,所述增塑剂包括邻苯二甲酸衍生物、己二酸衍生物、油酸衍生物、马来酸衍生物、硬脂酸衍生物中的任意一种或两种以上。其中,邻苯二甲酸衍生物例如邻苯二甲酸二甲酯、邻苯二甲酸二乙酯、邻苯二甲酸二异丁酯、邻苯二甲酸二己酯、邻苯二甲酸丁基乙癸酯;己二酸衍生物例如己二酸二正丁酯、己二酸二异丁酯、己二酸二异辛酯;油酸衍生物例如油酸丁酯;马来酸衍生物例如马来酸二正丁酯、马来酸二乙酯;硬脂酸衍生物例如硬脂酸正丁酯、硬脂酸甘油酯。
[0062]
第二方面,本发明提供了上文所述硬掩膜组合物的制备方法,所述制备方法为:取所述聚合物与所述溶剂混合,所述聚合物的质量占比为4%~25%,所述溶剂的质量占比75~95.6%。
[0063]
根据本发明的一些实施例,所述制备方法为:取所述聚合物与所述溶剂、交联剂、催化剂、表面活性剂以及增塑剂混合,以所述硬掩膜组合物的总量计,所述聚合物的质量占比为4%~25%,所述交联剂的质量占比0.4~3%,所述催化剂的质量占比0.001%~0.05%,所述表面活性剂的质量占比0.01%~0.1%,所述增塑剂的质量占比0%~2.5%,所述溶剂的质量占比75~95.6%。
[0064]
第三方面,本发明提供了一种形成图案的方法,所述方法包括以下步骤:在基板上提供材料层;将如上所述的硬掩膜组合物施加在所述材料层上;热处理所述硬掩膜组合物以形成硬掩膜;在所述硬掩膜上形成含硅薄层;在所述含硅薄层上形成光刻胶抗蚀层;将所述光刻胶抗蚀层曝光并显影以形成光刻胶图案;使用所述光刻胶图案选择性去除所述含硅薄层和所述硬掩膜以暴露所述材料层的一部分;刻蚀所述材料层的暴露部分。
[0065]
根据本发明的一些实施例,所述基板包括硅晶片、玻璃衬底或聚合物衬底中的任意一种。
[0066]
根据本发明的一些实施例,所述材料层为待最终图案化的材料,包括铝层或铜层等金属层、硅层等半导体层、二氧化硅或氮化硅等绝缘层。
[0067]
根据本发明的一些实施例,其中形成硬掩膜的方法为:将所述硬掩膜组合物以溶液形式旋涂在所述材料层上,在200℃到500℃下对所述硬掩膜组合物进行热处理,热处理时间约为10秒到10分钟,以形成硬掩膜。进一步的,所述硬掩膜组合物的厚度不受特定限制,可为100到10000埃米。
[0068]
根据本发明的一些实施例,所述含硅薄层包括氮化硅、氧化硅或氮氧化硅中的任意一种。
[0069]
根据本发明中的一些实施例,所述光刻胶抗蚀层曝光可使用例如arf、krf或euv进行光刻胶层的曝光。
[0070]
下面通过一些具体实施例对本发明作进一步说明。
[0071]
实施例1
[0072]
单体1的合成:
[0073]
在氩气保护下,向1000ml反应瓶中加入18.0g(50mmol)1,6-二溴芘、26.2g(105mmol)2-硝基苯硼酸频哪醇酯、27.6g(200mmol)碳酸钾、2.3g(2mmol)四三苯膦基钯和500ml甲苯,搅拌均匀后升温至120℃反应6h,冷却反应液,加水猝灭反应。分出有机相,水相使用甲苯萃取三次后合并有机相,用无水硫酸镁干燥,过滤,旋干溶剂,经硅胶薄层色谱提纯得到中间产物。
[0074]
在氩气保护下,向500ml反应瓶中加入17.8g(40mmol)中间产物、50.4g(192mmol)三苯基膦、0.69g(2mmol)钼基催化剂、1,2-二氯苯250ml,搅拌均匀后回流反应12h,冷却反应液至室温,使用甲苯萃取,之后水洗,无水硫酸镁干燥,过滤,旋干溶剂,经硅胶薄层色谱提纯得到单体1,所述单体1的碳含量为88.4%。上述单体1的合成过程如反应式1所示。
[0075][0076]
单体2的合成
[0077]
在氩气保护下,向500ml反应瓶中加入11.4g(30mmol)单体1,13.3g(65mmol)碘苯,9.0g(65mmol)碳酸钾,0.3g(0.6mmol)二(三叔丁基膦)钯,甲苯200ml,回流反应8h,之后将反应液冷却至室温,过滤,水洗,无水硫酸镁干燥,旋干溶剂,通过柱层析纯化,得到单体2,单体2的碳含量90.2%。
[0078]
单体2的合成过程如反应式2所示。
[0079][0080]
合成聚合物:
[0081]
聚合物1-1的合成
[0082]
向100ml三口烧瓶中加入3.80g(0.01mol)单体1,2.42g(0.01mol)3,5-二甲氧基甲基联苯和50ml丙二醇单甲醚乙酸酯,混合均匀后加入0.19g(0.001mol)对甲苯磺酸,在氮气保护下,100℃反应10h。反应结束后,冷却反应液,然后将反应液倒入甲醇中,以除去未反应的单体1和低分子量聚合物,过滤混合液,将滤饼进一步用甲醇清洗2次后,使用50℃真空烘箱干燥12h得到如化学式1-1表示重复单元的聚合物1(mw=4900,多分散度=2.1)。
[0083][0084]
实施例2
[0085]
重复实施例1的合成步骤,区别在于,使用2.42g(0.01mol)4,4
’‑
二甲氧基甲基联苯替代2.42g(0.01mol)3,5-二甲氧基甲基联苯,得到如化学式1-2表示重复单元的聚合物2(mw=5500,多分散度=1.8)。
[0086][0087]
实施例3
[0088]
重复实施例1的合成步骤,区别在于,使用2.58g(0.01mol)4,4
’‑
双甲氧基甲基-二苯醚替代2.42g(0.01mol)3,5-二甲氧基甲基联苯,得到如化学式1-3表示重复单元的聚合物3(mw=4400,多分散度=1.9)。
[0089][0090]
实施例4
[0091]
重复实施例3的合成步骤,区别在于,使用5.32g(0.01mol)单体2替代3.80g(0.01mol)单体1,得到如化学式1-4表示重复单元的聚合物3(mw=5900,多分散度=2.2)。
[0092][0093]
对比例4
[0094]
重复实施例1的合成步骤,区别在于,使用2.02g(0.01mol)芘替代单体1,使用1.66g(0.01mol)1,4
’‑
二甲氧基甲基苯替代2.42g(0.01mol)3,5-二甲氧基甲基联苯,得到如化学式1-5表示重复单元的聚合物5(mw=6700,多分散度=1.6)。
[0095][0096]
实施例5
[0097]
分别实施例1~4以及对比例1中制备得到的聚合物1~5,与溶剂、交联剂、催化剂和表面活性剂按照表1的配比混合均匀,得到抗蚀下层膜组合物1~5。其中,溶剂为丙二醇单甲醚乙酸酯(pgmea)和环己酮按照体积比7:3配制的混合溶剂,催化剂为对甲苯磺酸,表面活性剂为聚氧乙烯月桂基醚,交联剂为结构如下所示的三嗪化合物:
[0098][0099]
表1(wt%)
[0100][0101]
测试:
[0102]
通过下述的评价方法评价实施例和对比例的抗蚀下层膜组合物1~5的溶解性、空隙填充特性、平坦化特性和耐刻蚀性评价。
[0103]
(1)溶解性评价
[0104]
将合成的聚合物1、聚合物2、聚合物3、聚合物4、聚合物5分别以质量比15%的比例添加至pgmea中,在50℃下加热搅拌1小时后,确认溶解状态;之后冷却至25℃,立即确认溶解状态;最后,25℃静置6h,确认溶解状态,所得结果如表2所示。
[0105]
溶解性判定标准:
[0106]
◎
:25℃静置6h后,肉眼没有确认到未溶物;
[0107]
○
:加热状态和刚冷却状态下肉眼未确认到不溶物,但25℃静置6h后,肉眼确认到少量不溶物;
[0108]
△
:加热状态下肉眼未确认到不溶物,但在刚冷却状态下以及25℃静置6h后,肉眼确认到不溶物;
[0109]
表2
[0110]
项目溶解性聚合物1
○
聚合物1
○
聚合物1
◎
聚合物1
○
聚合物5
△
[0111]
从表2的结果可以看出,聚合物1~聚合物5,由于包含柔性的结构单元,表现出良好的溶解性能,而聚合物5由于分子刚性比较大,导致溶解性能比较差。
[0112]
(2)空隙填充特性和平坦化特性
[0113]
在线/间隔的宽度为160nm/200nm、高度为300nm的被覆了氧化硅的硅基板上,分别旋涂表1中制备得到的硬掩膜组合物1~5,在350℃进行烧成180s,形成抗蚀下层膜。利用场发射扫描电镜观察所形成的抗蚀剂下层膜的剖面,确定是否存在空隙,来评价间隙填充特性;
[0114]
再通过旋涂抗蚀下层膜的厚度高的部分(线部分)与厚度低的部分(间隔部分)之间的差来判定平坦化特性,其中,两者之差小于20nm则判定平坦化特性“非常优良”,在20nm-50nm之间则判定为“优良”,大于50nm则判定为“不良”,所得结果如表3所示。
[0115]
(3)耐刻蚀性评价
[0116]
将表1中实施例和对比例的硬掩膜组合物1~5,分别使用旋涂机器涂布在硅晶片上,在350℃进行180s烧成,形成厚度为250nm的抗蚀下层膜。之后使用cf4气体作为刻蚀气体进行干刻蚀并测定干刻蚀速率;此外,将0.5g市售的甲酚酚醛树脂(重均分子量5000,多分散度1.7)、0.05g与对比例相同的交联剂、0.005g对甲苯磺酸溶于4.5g pgmea所形成的溶液旋涂在硅基板上,形成硬掩膜,亦用cf4气体作为刻蚀气体进行干刻蚀并测定干刻蚀速率,与由实施例和比较例形成的硬掩膜1~5的干刻蚀速率进行比较得到干刻蚀速率比,将结果列于表3。表3中的干刻蚀速率比由公式1计算得到。
[0117]
公式1:
[0118]
干刻蚀速率比=硬掩膜1~5的干刻蚀速率/甲酚酚醛树脂的干刻蚀速率
[0119]
表3
[0120]
项目平坦化特性填隙特性干刻蚀速率比实施例1优良没有空隙0.71实施例2优良没有空隙0.70实施例3非常优良没有空隙0.70实施例4优良没有空隙0.69对比例1不良有空隙0.79
[0121]
从表3的测试结果可以看出,一方面,本发明提供的硬掩膜组合物因为具有优异的溶解性能,所以表现出优异的间隙填充特征和平坦化特征;另一方面,与对比例相比,本发明提供的硬掩膜组合物亦表现出改善的耐刻蚀性能。
[0122]
除非另作定义,本发明中使用的技术术语或者科学术语应当为本发明所属领域内具有一般技能的人士所理解的通常意义。本发明中使用的“第一”、“第二”以及类似的词语并不表示任何顺序、数量或者重要性,而只是用来区分不同的组成部分。“连接”或者“相连”等类似的词语并非限定于物理的或者机械的连接,而是可以包括电性的连接,不管是直接的还是间接的。“上”、“下”、“左”、“右”等仅用于表示相对位置关系,当被描述对象的绝对位置改变后,则该相对位置关系也相应地改变。
[0123]
以上所述是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明所述原理的前提下,还可以作出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。