1.本发明涉及医疗产品领域,且特别涉及一种荧光靶向纳米药物载体和靶向药物的制备方法。
背景技术:
2.胰腺癌在近些年来发病率逐年递增,并且由于它的不敏感性,目前临床上的治疗方案并没有达到预期效果。中医药在肿瘤免疫治疗中有独特优势。蟾酥是著名的民间中药,因其显著的抗肿瘤活性而备受关注。蟾毒灵是中药蟾酥的主要活性成分,现代药理学研究表明其对多种癌症具有抗肿瘤作用。由于蟾毒灵在血液、组织中的代谢,导致进入肿瘤部分的药物较少,治疗效果不明显。目前蟾毒灵存在的载药方式单一,药物利用度低,无法体内评估等问题。
技术实现要素:
3.本发明的目的是针对现有技术不足,提供了一种荧光靶向纳米药物载体和靶向药物的制备方法。
4.为实现上述发明目的,本发明采用以下技术方案来实现:一种荧光靶向纳米药物载体的制备方法,具体包括以下步骤:
5.(1)制备单分散中孔生物活性玻璃bgn;
6.(2)制备钙钛矿量子点;
7.(3)将步骤(1)制得的单分散中孔生物活性玻璃bgn与步骤(2)制得的钙钛矿量子点在室温条件下混合至在紫外灯下显绿色,并经过真空干燥箱室温挥发干燥后制得纳米颗粒 bgn-qds;
8.(4)制备荧光靶向纳米药物载体:将步骤(3)制备的纳米颗粒bgn-qds作为荧光团并用叶酸官能化,具体为:首先在80℃下用(3-氨基丙基)三乙氧基硅烷将步骤(3)制得的纳米颗粒bgn-qds胺化过夜,所述纳米颗粒bgn-qds与(3-氨基丙基)三乙氧基硅烷的质量体积比为2~5mg/ml,得到浓度为4~9mg/ml的胺化后的bgn-qds混合物溶液,作为荧光团;将浓度为0.15m的1-乙基-3-(3-二甲基氨基丙基)碳二亚胺在2-8℃下溶解在 300~600μg/ml叶酸溶液中;此后,将胺化后的bgn-qds混合物溶液中在室温搅拌均匀,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将该溶液离心,洗涤,然后置于冷冻环境下冷冻12h~36h后,冻干除去水分,得到荧光靶向纳米药物载体。
9.进一步地,所述步骤(1)制备单分散中孔生物活性玻璃bgn具体为:先将十二烷基胺溶解于20ml去离子水和80ml无水乙醇中,得到浓度为0.02~0.08m的十二烷基胺溶液, 当十二烷基胺完全溶解后,在磁力搅拌下,加入8-16ml正硅酸四乙酯后,在30~50℃下反应30~60分钟,加入0.61ml-1.22ml磷酸三乙基酯和1.685g-3.39g硝酸钙四水合物,充分搅拌,反应完全后,形成白色沉淀;过滤,收集白色沉淀物,洗涤该白色沉淀物,冷冻干燥后;按升温速率2℃/min升温至700℃,然后煅烧两小时,炉内冷却过夜胺化,即得。
10.进一步地,所需十二胺浓度优选为0.08m;所需正硅酸四乙酯溶液优选为16ml;所述磷酸三乙基酯优选为1.22ml,所述硝酸钙四水合物优选为3.39g;
11.进一步地,所述步骤(4)中冷冻环境温度优选低于-20℃;纳米颗粒bgn-qds与(3
‑ꢀ
氨基丙基)三乙氧基硅烷的质量体积比优选为5mg/ml。
12.一种荧光靶向纳米药物载体,上述的方法制备得到。
13.本发明提供了一种靶向药物的制备方法具体为:在300~600μg/ml过量的叶酸溶液中,将荧光靶向纳米药物载体与抗肿瘤药物按质量比150-200:1混合,离心过滤,得到靶向药物。
14.进一步地,所述抗肿瘤药物优选抗肿瘤药物dox或抗肿瘤中药单体。
15.进一步地,所述抗肿瘤中药单体优选浓度为0.01mg/ml~0.03mg/ml的蟾毒灵。
16.进一步地,所述抗肿瘤中药单体优选浓度为0.02mg/ml的蟾毒灵。
17.一种靶向药物,由上述制备方法制备得到。
18.本发明的有益效果为:本发明构建了“荧光协同”策略介导的蟾毒灵靶向制剂,针对体内药物评估困难,利用材料科学和生命科学的结合,从包载、组装及其体内可视化评估的研究,通过“核壳结构”构建并完善了bgn和qds的共载,基于此改进,防止量子点的荧光猝灭,并降低细胞毒性,使其成为肿瘤可视化评估的识别基团。同时,利用中药学和有机化学原理及生物学研究手段,引进靶向配体—叶酸和抗肿瘤中药单体—蟾毒灵,设计、合成“荧光协同”策略介导的蟾毒灵靶向制剂并进行构效关系研究,实现肿瘤的靶向给药,通过可视化评估载体在肿瘤部位的聚集程度以及药物的靶向能力。开展材料学结构、生物学以及抗肿瘤免疫作用机制的研究和验证。克服了蟾毒灵现有的局限性,为新型中药载体的设计、开发和安全应用提供理论基础,同时为肿瘤动态可视化提供了技术支撑。
附图说明
19.为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
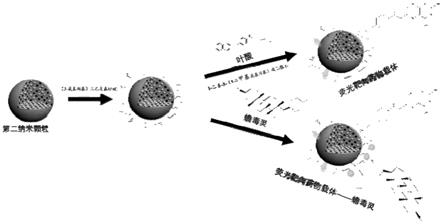
20.图1为本发明荧光靶向药物载体合成路线图;
21.图2为本发明实施例1、实施例2、实施例3所述纳米颗粒的x-射线能谱图;
22.图3为本发明实施例1、实施例2、实施例3提供的不同孔径bgn——量子点的荧光强度示意图;
23.图4为本发明实施例1所示单纯量子点以及bgn-量子点的时间-荧光强度示意图;
具体实施方式
24.为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
25.下面对本发明实施例的荧光靶向纳米药物载体及其制备方法进行具体说明。
26.本发明提供一种荧光靶向药物载体的制备方法,荧光靶向药物载体合成路线图如
图1所示,其包括:
27.(1)制备不同孔径的单分散中孔生物活性玻璃(bgn):使用改进的溶胶-凝胶法优化生物活性玻璃(bgn)合成配比,使用过十二烷基胺(dda)作为催化剂和有机模板剂合成。具体操作如下:先将的将十二烷基胺(dda)溶解于20ml去离子水(dw)和80ml无水乙醇(etoh)中,得到浓度为0.02~0.08m的十二烷基胺溶液,当dda完全溶解后,在磁力搅拌下,加入8-16ml正硅酸四乙酯(teos)后,在30~50℃下反应30~60分钟,加入0.61ml-1.22ml磷酸三乙基酯(tep)和1.685g-3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,反应完全后,形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水多次洗涤,冷冻干燥后。之后通过700
°
以2℃ /min煅烧两小时,炉内冷却过夜,炉内冷却过夜胺化,即得到不同孔径的bgn,并于室温保存。
28.所述正硅酸四乙酯、磷酸三乙基酯(tep)和硝酸钙四水合物比例优选为16ml:1.22ml: 3.39g。
29.(2)制备钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
30.(3)将步骤(1)制得的单分散中孔生物活性玻璃(bgn)与步骤(2)制得的钙钛矿量子点在室温条件下混合至在紫外灯下显绿色,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
31.(4)制备荧光靶向纳米药物载体和靶向药物:
32.所述制备荧光靶向纳米药物载体的步骤具体为:将步骤(3)制备的纳米颗粒bgn-qds作为荧光团并用叶酸官能化。具体为首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes) 将步骤(2)制得的纳米颗粒bgn-qds胺化过夜,所述纳米颗粒bgn-qds与(3-氨基丙基) 三乙氧基硅烷(aptes)的质量体积比为2~5mg/ml,得到浓度为4~9mg/ml的胺化后的 bgn-qds混合物溶液,作为荧光团。之后,将浓度为0.15m的1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在2-8℃浓度为300~600μg/ml的叶酸溶液中。此后,将胺化后的bgn-qds混合物溶液中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds 混合物过量,并将该溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于冷冻环境下冷冻12h~36h后,冻干除去水分,得到荧光靶向纳米药物载体。其中,本发明较佳的实施例中,冷冻环境低于-20℃,冻干效果更佳。
33.所述制备靶向药物的步骤具体为:在300~600μg/ml过量的叶酸溶液中,步骤(4)制得的荧光靶向纳米药物载体与抗肿瘤药物按质量比150-200:1混合,得到靶向药物。所述抗肿瘤药物优选抗肿瘤药物dox或抗肿瘤中药单体,所述抗肿瘤中药单体优选蟾毒灵。
34.本发明较佳的实施例中,可以选取抗肿瘤药物dox,抗肿瘤中药单体等等,所述抗肿瘤中药单体优选蟾毒灵。
35.本发明较佳的实施例中,蟾毒灵浓度为0.01mg/ml~0.03mg/ml,优选浓度为0.02mg/ml 的蟾毒灵。
36.本发明提供一种新型荧光靶向药物载体,并结合中药单体——蟾毒灵,其由上述荧光靶向药物载体的制备方法制得,其具有较佳的荧光性能以及抗肿瘤性,可作为肿瘤的
靶向定位以及治疗。
37.本发明实施例的一种新型荧光靶向纳米药物载体及其制备方法的有益效果是:本发明方法克服了蟾毒灵现有的局限性,为新型中药载体的设计、开发和安全应用提供理论基础,同时为肿瘤动态可视化提供了技术支撑。可作为肿瘤的靶向治疗载体。
38.以下结合实施例对本发明的特征和性能作进一步的详细描述。
39.实施例1
40.本发明提供一种新型荧光靶向药物载体的制备方法,其包括荧光单体、靶向载体和抗肿瘤药物,荧光单元是钙钛矿量子点,靶向纳米颗粒载体由纳米颗粒制备,抗肿瘤药物选取中药单体——蟾毒灵。
41.一种新型荧光靶向药物载体由以下方法制得:
42.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中当dda完全溶解后,得到浓度为0.02m的十二胺溶液,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得到bgn。
43.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilization and easy phase transfer of cspbbr3 perovskite quantum dots promoted by highaffinity polyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–ꢀ
12680.》所示的方法合成钙钛矿量子点。
44.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
45.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将2mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
46.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
47.图4为本发明实施例1所示单纯量子点以及bgn-量子点的时间-荧光强度示意图;由图 4可得,本发明实施例1制得的bgn-量子点随时间增大其荧光强度远高于单纯量子点。量子点存在猝灭现象,随着时间变化,该现象越发明显,然而bgn-量子点通过核壳结构保护了量子点,使得它猝灭更慢,该实验量化了时间。
48.实施例2
49.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.04m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将 16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不
透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,再煅烧两小时,炉内冷却过夜,即得bgn。
50.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
51.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
52.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将2mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
53.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
54.实施例3
55.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将 16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得到bgn。
56.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
57.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
58.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将2mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ml 冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤2、3、5或7次等,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
59.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
60.表1:本发明实施例1、实施例2、实施例3制得的纳米颗粒表
61.dda浓度比表面积(m2/g)颗粒孔径(cm3/g)颗粒大小(nm)
0.02294.19031.624542108.4070.04300.06210.843065145.8810.08287.37241.086068212.788
62.图2为本发明实施例1、实施例2、实施例3所述纳米颗粒的x-射线能谱图;x-射线能谱图用于观测晶型,量子点存在晶型,通过该方式,用于检测bgn-qds中量子点的存在。如图所示,量子点的特征峰如图谱pdf54-0752,bgn-qds在对应角度有个峰高,证明其存在量子点。作为对照,bgn则不存在峰高。
63.图3为本发明实施例1、实施例2、实施例3提供的不同孔径bgn——量子点的荧光强度示意图,孔径越大能检测到的量子点荧光强度越高。
64.由上表1、图2和图3可知,十二胺(dda)的浓度优选为0.08m。此时,制备得到的生物活性玻璃bgn颗粒大小最大,荧光强度最强,反应效果最佳。
65.实施例4
66.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇(etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后通过700
°
以2℃/min煅烧两小时,炉内冷却过夜,即得bgn。
67.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilizationandeasyphasetransferofcspbbr3perovskitequantumdotspromotedbyhighaffinitypolyzwitterionicligands,”in2020.am.chem.soc.,jun2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
68.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
69.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将2.5mg/ml的bgn-qds胺化过夜。之后,将0.15m1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
70.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的bufalin与2mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
71.实施例5
72.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇(etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
73.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilization and easy phase transfer of cspbbr3 perovskite quantum dots promoted by highaffinity polyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–ꢀ
12680.》所示的方法合成钙钛矿量子点。
74.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
75.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将3mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,并将溶液离心,用dw洗涤,例如用去离子水洗涤2、3、5或7次等,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
76.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.02mg/ml的bufalin与1.8mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
77.实施例6
78.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m的十二胺,当dda完全溶解后,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
79.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilization and easy phase transfer of cspbbr3 perovskite quantum dots promoted by highaffinity polyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–ꢀ
12680.》所示的方法合成钙钛矿量子点。
80.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
81.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将3.5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
82.s5.制备靶向药物:在500μg/ml过量的叶酸溶液中,将0.01mg浓度为0.02mg/ml的 bufalin与2mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
83.实施例7
84.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m十二胺溶液,当dda完全溶解后,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸
钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
85.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilizationandeasyphasetransferofcspbbr3perovskitequantumdotspromotedbyhighaffinitypolyzwitterionicligands,”in2020.am.chem.soc.,jun2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
86.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
87.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将4mg/ml的bgn-qds胺化过夜。之后,将0.15m1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
88.s5.制备靶向药物:在600μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
89.实施例8
90.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇(etoh)中当dda完全溶解后,得到浓度为0.08m的十二胺溶液,在磁力搅拌下,将16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
91.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilizationandeasyphasetransferofcspbbr3perovskitequantumdotspromotedbyhighaffinitypolyzwitterionicligands,”in2020.am.chem.soc.,jun2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
92.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
93.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ml冰冷叶酸溶液中。此后,将胺化后的4mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
94.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
95.实施例9
96.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将 16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
97.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
98.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
99.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的4.5mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
100.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
101.实施例10
102.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将 16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
103.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilization and easy phase transfer of cspbbr3 perovskite quantum dots promoted by highaffinity polyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–ꢀ
12680.》所示的方法合成钙钛矿量子点。
104.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
105.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的5mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃,冷冻环境下冷冻12h-36h后,冻干除去水分。
106.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml 的bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
107.实施例11
108.s1.制备生物活性玻璃:先将的十二胺溶解于20ml去离子水(dw)和80ml无水乙醇(etoh)中当dda完全溶解后,得到得到浓度为0.08m的十二胺溶液,在磁力搅拌下,将16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
109.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
110.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
111.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的5.5mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
112.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml 的bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
113.实施例12
114.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中当dda完全溶解后,得到浓度为0.08m的十二胺溶液,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
115.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
116.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
117.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸
溶液中。此后,将胺化后的6mg/ml的bgn-qds混合物中在室温搅拌24小时,并将溶液离心,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
118.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
119.实施例13
120.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
121.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
122.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
123.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的6.5mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
124.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
125.实施例14
126.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中,得到浓度为0.08m的十二胺溶液,当dda完全溶解后,在磁力搅拌下,将 16ml正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep)和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
127.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
128.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
140.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
141.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的8mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
142.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
143.实施例17
144.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中当dda完全溶解后,得到浓度为0.08m的十二胺溶液,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
145.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
146.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
147.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ml 冰冷叶酸溶液中。此后,将胺化后的8.5mg/ml的bgn-qds混合物中在室温搅拌24小时,并将溶液离心,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
148.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的 bufalin与1.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
149.实施例18
150.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中当dda完全溶解后,得到浓度为0.02m的十二胺溶液,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
151.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
152.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
153.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的9mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
154.s5.制备靶向药物:在300μg/ml过量的叶酸溶液中,将0.01mg浓度为0.01mg/ml的bufalin与2mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
155.实施例19
156.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中当dda完全溶解后,得到浓度为0.08m的十二胺溶液,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
157.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhanced stabilizationand easy phase transfer of cspbbr3 perovskite quantum dots promoted by high affinitypolyzwitterionic ligands,”in 2020.am.chem.soc.,jun 2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
158.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
159.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m 1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ ml冰冷叶酸溶液中。此后,将胺化后的9mg/ml的bgn-qds混合物中在室温搅拌24小时,所述叶酸溶液相对于胺化后的bgn-qds混合物过量,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
160.s5.制备靶向药物:在500μg/ml过量的叶酸溶液中,将0.02mg浓度为0.02mg/ml的 bufalin与3mg荧光靶向纳米药物载体混合,即得到bgn-qds-fa-bufalin。
161.实施例20
162.s1.制备生物活性玻璃:先将十二胺溶解于20ml去离子水(dw)和80ml无水乙醇 (etoh)中当dda完全溶解后,得到浓度为0.08m的十二胺溶液,在磁力搅拌下,将16ml 正硅酸四乙酯(teos)加入后,在40℃下反应30分钟,加入1.22ml磷酸三乙基酯(tep) 和3.39g硝
酸钙四水合物(cn)继续剧烈搅拌所得溶液3小时,由于形成白色沉淀,澄清溶液逐渐变成不透明。然后,通过过滤收集白色沉淀物,用无水乙醇和去离子水冲洗三次,冷冻干燥后。之后按升温速率2℃/min升温至700℃,煅烧两小时,炉内冷却过夜,即得bgn。
163.s2.制备所述钙钛矿量子点;采用《s.s.wang,l.du,z.c.jin,y.xin,“enhancedstabilizationandeasyphasetransferofcspbbr3perovskitequantumdotspromotedbyhighaffinitypolyzwitterionicligands,”in2020.am.chem.soc.,jun2020,pp.142,29,12669
–
12680.》所示的方法合成钙钛矿量子点。
164.s3.将所述的单分散中孔生物活性玻璃与钙钛矿量子点在室温条件下混合,并经过真空干燥箱室温挥发干燥后制得纳米颗粒bgn-qds;
165.s4.制备荧光靶向纳米药物载体:将制备的bgn-qds作为荧光团并用叶酸官能化。为此,首先在80℃下用(3-氨基丙基)三乙氧基硅烷(aptes)将5mg/ml的bgn-qds胺化过夜。之后,将0.15m1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(edac)溶解在500μg/ml冰冷叶酸溶液中。此后,将胺化后的9mg/ml的bgn-qds混合物中在室温搅拌24小时,并将溶液离心,用dw洗涤,例如用去离子水洗涤多次,然后置于-20℃冷冻环境下冷冻12h-36h后,冻干除去水分。
166.s5.制备靶向药物:在600μg/ml过量的叶酸溶液中,将0.03mg浓度为0.01mg/ml的bufalin与4.5mg荧光靶向纳米药物载体混合,离心过滤,即得到bgn-qds-fa-bufalin。
167.综上,本发明实施例提供的新型荧光靶向药物载体的制备方法,通过该方法可以合成可控粒径的荧光靶向药物载体,可以实现体内靶向作用以及肿瘤治疗。本发明提供的新型荧光靶向药物载体,其具有较佳的荧光性能以及靶向性,可作为肿瘤的治疗以及动态评估剂。
168.以上所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。