1.本发明属于测序技术领域,具体涉及一种制备二代测序接头的方法。
背景技术:
2.接头是二代文库的必要组成部分。它一般由测序平台特异性的已知dna序列组成,通常包括3大功能成分:通用pcr引物结合位点(pcr用来做文库富集)、样本index(在多样本混样测序中用来对测序的read进行拆分)以及测序引物结合位点(用来与测序引物进行退火)。随着ngs技术应用的拓展,接头的结构也在不断演化。如对低频ctdna突变的检测,为了克服ngs技术自身的一些缺点,如针对在使用pcr构建文库和测序过程中自身发生的错误,通常会在文库构建时引入独特的分子标签(unique molecular identifier,umi),即在每个dna分子扩增前都加上一个umi。扩增后,具有相同umi的dna序列被视为来源于同一个dna分子,这样可达到dna分子的精确定量。同时,由于文库构建时pcr以及测序过程产生的错误是随机的,在对具有相同umi的dna序列进行一致性的整合后,可消除这些随机产生的错误。
3.目前,不管是常规接头,还是包含umi的接头,生产和制备最常用的方法是通过人工合成部分互补的寡聚核苷酸后进行退火形成。此方法通常是通过合成2条寡具核苷酸进行退火形成接头(比如wo 2018/148289 a2,us2019/0085384 a1等专利公开的方法),但由于其5’端磷酸基团是由化学修饰添加的,添加效率小于100%,没有5’端磷酸基团将不能完成链接反应,从而对后续的链接效率产生负面的影响。在其它接头制备方法中,用得最多的是通过酶学方法制备接头(比如cn111471746a,cn109402224a等专利公开的方法),该方法的反应体系中包括模板和引物,模板中包含简并碱基作为umi,并且与引物可退火的寡聚核苷酸长度较短(一般长度为16bp,退火温度在50摄氏度左右),导致退火温度较低,在较低的退火温度下,这些简并碱基极有可能与模板中的其它固定序列进行配对结合,或形成二级结构,或与体系中的引物序列进行配对,导致引物结合到错误的模板上,致使在下一步的链延伸过程中产生错误的产物,从而使得引物利用率降低,最终导致生成正确接头的效率降低,而且这种方法的步骤较多,操作复杂。因此,有必要研发新型的接头制备方法,以提高产生有效接头的效率。
技术实现要素:
4.为了克服上述现有技术的不足,本发明提出了一种制备二代测序接头的方法,本发明方法制备过程更为简洁,而且产生有效接头的效率更高。
5.为了实现上述目的,本发明所采用的技术方案是:
6.本发明提供了一种制备二代测序接头的方法,具体为:根据测序平台选择两条可互补序列,所述可互补序列由接头序列,限制性酶切位点序列和用于增加退火温度的序列组成,两条可互补序列的长度均大于16bp,先对两条可互补序列进行退火处理,然后使用能产生t凸出或平末端的限制性内切酶进行酶切消化,消化后对接头进行纯化即得。
7.一方面,本发明接头的制备由酶反应完成,5’端磷酸基团由酶切水解产生,只要水
解完成,将更有效的产生有效的5’端磷酸基团,从而可以提高接头的链接效率。另一方面,本发明采用了2大创新性设计:(1)接头互补的模板序列中,采用了更长的可互补序列(大于116bp,本发明使用了49bp的互补序列,其退火温度大于70℃),意味着互补的模板可在更高的温度中进行退火,降低简并碱基模板中的其它固定序列的退火几率,从而提高正确接头的生成率;(2)与现有的接头制备方法(比如cn111471746a,cn109402224a)相比,退火后的接头模板直接被酶切消化,而不需要先进行额外的链延伸反应或链延伸反应后的纯化步骤。因为链延伸反应属于酶学反应,效率不能达到100%,会导致生成有效接头的效率进一步降低。
8.优选地,用于增加退火温度的序列长度大于1bp,小于50bp,gc含量大于40%。
9.优选地,所述接头序列可包含双端index,或单端index,或双端umi,或单端umi,可在位于兼并碱基处进行设计,也可以使用测序平台特异性接头的部分序列,且部分序列中不包含umi或/和index,在此情况下umi和/或index可在富集pcr引物中添加。
10.进一步地,所述接头序列包括适用于illumina测序平台的第一序列和第二序列,所述第一序列如seq id no:1所示,所述第二序列如seq id no:2所示。
11.优选地,退火的体系为:两条互补接头序列的浓度均为100微摩,体积均为5微升;退火的程序为:95℃退火2分钟;然后以0.1℃/秒的速度降温至60℃。
12.优选地,所述限制性内切酶包括xcmi,ahdi,bcivi,bmri,fnu4hi,hphi,hpy188i,mboii,mnli,bsabi。进一步地,所述限制性内切酶为xcmi。
13.产生的接头可以是含t凸出,用于进行t/a链接。也可是产生平末端。可通过在模板中使用不同的限制性酶切位点来实现。比如使用bsabi酶切位点,则可以产生带umi序列平末端接头。
14.优选地,酶切消化的总体积为30μl,包括10μm退火产物10μl,无核酸酶水14μl,10
×
cutsmarter缓冲液3μl,5u/μl限制性内切酶3μl;酶切消化的反应程序为37℃孵育2小时。
15.优选地,纯化方法包括链霉亲和素磁珠吸附法。也可以使用其它可分离dna寡具核苷酸的方法,比如page,柱纯化方法。
16.与现有技术相比,本发明的有益效果是:
17.本发明提供了一种制备二代测序接头的方法,该方法中的接头由酶切消化产生,相比于合成方法制备的接头,将产生更多有效的5’端磷酸基团,可以提高接头的链接效率;同时,本发明采用长模板退火,降低退火过程中简并碱基与其它固定序列(分子内或分子间)的结合,提高接头生成效率;次外,退火后直接消化,避免了其它方法中(如酶学方法)由于在链延伸步骤因为效率问题而导致的接头生成效率降低。可见,本发明采用酶切消化的方法制备接头,不仅制备过程更为简洁,而且产生有效接头的效率更高。
附图说明
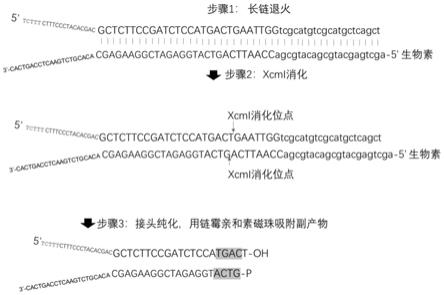
18.图1为接头制备的流程图;
19.图2为纯化后的接头与dna链接后形成的文库结构;
20.图3为制备的文库片段大小分布图。
具体实施方式
21.下面对本发明的具体实施方式作进一步说明。在此需要说明的是,对于这些实施方式的说明用于帮助理解本发明,但并不构成对本发明的限定。此外,下面所描述的本发明各个实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互组合。
22.下述实施例中的实验方法,如无特殊说明,均为常规方法,下述实施例中所用的试验材料,如无特殊说明,均为可通过常规的商业途径购买得到。
23.实施例1 一种制备二代测序接头的方法(以illumina测序平台,xcmi内切酶的使用为例)
24.该接头制备方法的制备流程如图1所示,具有包括以下步骤:
25.(1)从dna引物合成供应商(金斯瑞生物科技股份有限公司)订购以下dna寡聚核苷酸(该寡聚核苷酸长度较长,为提高其纯度,使用聚丙烯酰胺电泳方法(page)纯化得到,page纯化方法是寡具核苷酸合成工业中需要较高纯度时使用的标准纯化方法):
26.>seq-1(第一序列)
[0027]27.(seq id no:1;带下划横线部分为接头序列;带下划波浪线部分为互补序列,带双下划横线部分为限制性酶切位点);
[0028]
》seq-2(第二序列)
[0029][0029]
(seq id no:2;带下划横线部分为接头序列;带下划波浪线部分为互补序列,带双下划横线部分为限制性酶切位点)。
[0030]
(2)按以下条件对上述第一序列(seq-1)与第二序列(seq-2)进行退火:
[0031]
试剂浓度(微摩)体积(微升)seq-11005seq-21005
[0032]
退火在pcr仪中进行,退火条件为:95℃退火2分钟;然后以0.1℃/秒的速度降温至60℃,得到退火产物。
[0033]
(3)对上述退火产物进行酶切消化反应,反应的体系如下表:
[0034]
试剂组分组分终浓度组分体积(μl)无核酸酶水 14cutsmarter缓冲液(neb)10
×
3退火产物10μm10xcmi限制性内切酶5u/μl3总体积 30
[0035]
反应在pcr仪中进行,反应条件为:37℃孵育2小时,反应结束后得到消化好的接头。
[0036]
(4)纯化消化好的接头
[0037]
本实施案例中,先使用链霉亲和素磁珠对含生物素的副产物进行吸附去除,再用苯酚-氯仿法提取上清液中的接头dna。具体操作如下:
[0038]
将酶切产物(30μl)加入到30μl的2
×
b&w buffer(thermo fisher scientific)重悬的t1磁珠(dynabeads
tm
myone
tm
链霉亲和素t1,thermo fisher scientific,cat:65601)中,涡旋混匀,轻微离心,室温旋转一个小时,取上清液用苯酚-氯仿法提取dna,最后用50μl eb(洗脱液)重悬dna,得到纯化后的接头。
[0039]
(5)接头建库测试
[0040]
接头测试通过待链dna末端修复和链接两步进行:
[0041]
1)待链dna通过以下方法制备:
[0042]
使用以下引物扩增人基因组上的一个217bp的片段,该片段在基因组(hg19)中的位置如下:
[0043]
》chr12:25398196-25398412 217bp
[0044]
tactggtggagtatttgatagtgtattaaccttatgtgtgacatgttctaatatagtcacattttcattatttttattataaggcctgctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatccaacaatagaggtaaatcttgtt(seq id no:3)。
[0045]
上游引物:5'tactggtggagtatttgatagtgta 3'(seq id no:4);
[0046]
下游引物:5'aacaagatttacctctattgttgg 3'(seq id no:5)。
[0047]
扩增按照下表所示的反应体系进行:
[0048][0049]
扩增条件为:95℃5min,(95℃15s,55℃30s,72℃30s)40循环,72℃5min,4℃保存。
[0050]
对pcr产物使用1.8
×
体积的spri磁珠(beckman coulter)进行纯化,并将纯化后的产物溶解在40μl无核酸酶水中备用。
[0051]
2)末端修复:对待链接片段(217bp的pcr产物,提前制备)进行末端修复,反应如下:
[0052]
试剂用量无核酸酶水(天根)14.67ul217bp的pcr产物(10ng/ul)2ulkapa end repair & a-tailing buffer(kapa)2.34μlkapa end repair & a-tailing enzyme mix(kapa)1.00μl总体积20μl
[0053]
在pcr仪中,运行如下程序:20℃,30min;65℃,30min,得到末端修复后的产物。
[0054]
3)末端修复反应完毕后,加上链接反应所需的试剂和纯化好的接头如下:
[0055][0056][0057]
反应如下:20℃,1小时;4℃,过夜,得到链接产物,如图2和图3所示。
[0058]
3)链接反应结束后,使用1
×
体积的spri磁珠(beckman coulter)对链接产物进行纯化,并将纯化后的产物溶解在10μl无核酸酶水中。使用qseq片段分析仪对片段进行分析。
[0059]
结果如图3所示,得到未链接dna,单端链接的dna以及双端链接的dna的片段分布以及荧光强度,其中,大部分产物为双端链接的dna产物。根据qseq对各个片段进行定量的结果,未链接dna浓度为3.54nm,单端链接dna浓度为0.15nm,双端链接dna浓度为7.04nm。算得链接效率为:7.04/(7.04+0.15+3.54)
×
100%=65%,高于一般情况下约30%的链接效率,表明使用本发明制备的接头是有效的。
[0060]
以上对本发明的实施方式作了详细说明,但本发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。