1.本发明涉及医药合成技术领域,具体涉及一种米诺地尔的制备方法。
背景技术:
2.米诺地尔别名长压定或敏乐定,化学名称为:6-(1-哌啶基)-2,4-嘧啶二胺-3-氧化物,呈白色或类白色结晶性粉末。临床上,米诺地尔作为钾离子通道开放剂,能直接松弛血管平滑肌,有强大的小动脉扩张作用,使外周阻力下降,血压下降,而对容量血管无影响,故能促进静脉回流。同时,由于反射性调节作用和正性频率作用,可使心输出量及心率增加,但不引起体位性低血压。
3.近年发现米诺地尔对于脂溢性脱发的有效率可以达到90%,对斑秃、原发性三角形脱发等多种类型的脱发都有较好的临床治疗效果。临床表明它可以直接刺激毛囊的增殖和分化,促使血管生成,增加局部血液供应,开放钾通道,使毛囊由休止期向生长期转化,并延长生长期。
4.大多数文献报道,米诺地尔由氰乙酸甲酯和硝基胍环合、氯化得到4-氯-2,6-二氨基嘧啶,再与过苯甲酸进行氧化反应获得6-氨基-1,2-二氢-1-羟基-2-亚氨基-4-氯嘧啶,最后与六氢吡啶缩合制得米诺地尔。该方法使用硝基胍作为原料,硝基胍属于炸药制备原料,极易发生爆炸等危险,不易工业化生产。
5.中国专利cn107129470a以2,4-二氨基-6-氯嘧啶为起始原料,经过氧苯甲酸氧化后,在碱催化剂和其他有机试剂作为溶剂条件下和哌啶缩合形成米诺地尔。该方法需要无机碱作为催化剂,引入了无机物,需要采用有机溶剂乙腈或1,4-二氧六烷作为溶剂才能让反应进行,得到的米诺地尔纯度和收率有待提高。
技术实现要素:
6.本发明的目的在于提供一种米诺地尔的制备方法,本发明能够提高米诺地尔的纯度和收率。
7.为了实现上述发明目的,本发明提供以下技术方案:
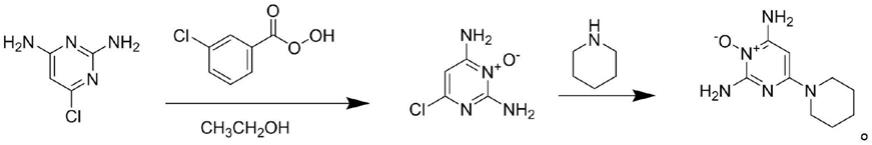
8.本发明提供了一种米诺地尔的制备方法,包括以下步骤:
9.将2,4-二氨基-6-氯嘧啶、间氯过氧苯甲酸和醇类溶剂混合,进行氧化反应,得到2,4-二氨基-6-氯嘧啶-1-氧化物;
10.将所述2,4-二氨基-6-氯嘧啶-1-氧化物和哌啶混合,进行氮氧化反应,得到米诺地尔。
11.优选地,所述2,4-二氨基-6-氯嘧啶和间氯过氧苯甲酸的摩尔比为1.0~2.0:1。
12.优选地,所述醇类溶剂包括乙醇、甲醇和异丙醇中的一种或多种。
13.优选地,所述氧化反应的温度为30~40℃;所述氧化反应的时间为3~8h。
14.优选地,所述2,4-二氨基-6-氯嘧啶-1-氧化物和哌啶的摩尔比为10~20:1。
15.优选地,所述氮氧化反应的温度为100~106℃;所述氮氧化反应的时间为3h以上。
16.优选地,所述氮氧化反应后还包括:将氮氧化反应所得米诺地尔粗品和精制溶剂混合,进行精制,得到精制体系;将所述精制体系依次进行析晶和抽滤,得到滤饼;将所述滤饼依次进行洗涤和干燥,得到米诺地尔。
17.优选地,所述精制溶剂包括异丙醇、甲醇、乙醇、丙酮和水中的一种或多种。
18.优选地,所述精制的温度为75~80℃。
19.优选地,所述析晶的温度为0~10℃。
20.本发明提供了一种米诺地尔的制备方法,包括以下步骤:将2,4-二氨基-6-氯嘧啶、间氯过氧苯甲酸和醇类溶剂混合,进行氧化反应,得到2,4-二氨基-6-氯嘧啶-1-氧化物;将所述2,4-二氨基-6-氯嘧啶-1-氧化物和哌啶混合,进行氮氧化反应,得到米诺地尔。本发明以2,4-二氨基-6-氯嘧啶和哌啶为起始原料,通过间氯过氧苯甲酸氧化形成氧化物后,与哌啶发生氮氧化反应形成目标化合物米诺地尔。本发明能够提高米诺地尔的纯度和收率。实施例结果表明,本发明制备的米诺地尔的纯度为99.81~100.0%,收率为85.1~86.2%。
具体实施方式
21.本发明提供了一种米诺地尔的制备方法,包括以下步骤:
22.将2,4-二氨基-6-氯嘧啶、间氯过氧苯甲酸和醇类溶剂混合,进行氧化反应,得到2,4-二氨基-6-氯嘧啶-1-氧化物;
23.将所述2,4-二氨基-6-氯嘧啶-1-氧化物和哌啶混合,进行氮氧化反应,得到米诺地尔。
24.在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
25.本发明将2,4-二氨基-6-氯嘧啶、间氯过氧苯甲酸和醇类溶剂混合,进行氧化反应,得到2,4-二氨基-6-氯嘧啶-1-氧化物。在本发明中,所述2,4-二氨基-6-氯嘧啶和间氯过氧苯甲酸的摩尔比优选为1.0~2.0:1。在本发明中,所述醇类溶剂优选包括乙醇、甲醇和异丙醇中的一种或多种。
26.在本发明中,所述2,4-二氨基-6-氯嘧啶、间氯过氧苯甲酸和醇类溶剂混合优选包括:将2,4-二氨基-6-氯嘧啶溶于部分醇类溶剂中,得到2,4-二氨基-6-氯嘧啶溶液;将间氯过氧苯甲酸溶于剩余醇类溶剂中,得到间氯过氧苯甲酸溶液;将所述间氯过氧苯甲酸溶液滴加至所述2,4-二氨基-6-氯嘧啶溶液中。在本发明中,所述2,4-二氨基-6-氯嘧啶和部分醇类溶剂的质量比优选为1:15~16,更优选为1:15.80。在本发明中,所述2,4-二氨基-6-氯嘧啶的溶解过程优选在搅拌条件下进行;溶解的温度优选为35~40℃。本发明优选将所述2,4-二氨基-6-氯嘧啶溶液的温度降至室温后再加入间氯过氧苯甲酸溶液。
27.在本发明中,所述间氯过氧苯甲酸和剩余醇类溶剂的质量比优选为1:3~5,更优选为1:3.94。
28.在本发明中,所述间氯过氧苯甲酸溶液的滴加速度优选为0.5l/min。本发明采用滴加的方式能够避免因一次性加入大量的间氯过氧苯甲酸造成的爆炸风险;同时也能够避免杂质产生,提高产品纯度。
29.在本发明中,所述氧化反应的温度优选为30~40℃;所述氧化反应的时间优选为3
~8h,更优选为5~6h。本发明优选采用tlc监测反应终点。
30.在本发明中,所述氧化反应后,优选将所得氧化反应体系进行纯化,得到2,4-二氨基-6-氯嘧啶-1-氧化物。在本发明中,所述纯化优选包括:向氧化反应体系中加入氢氧化钠,搅拌降温析晶后抽滤;将所得滤饼用无水乙醇洗涤,干燥后得到2,4-二氨基-6-氯嘧啶-1-氧化物。在本发明中,所述氢氧化钠优选以氢氧化钠固体形式加入或者以氢氧化钠水溶液形式加入;所述氢氧化钠水溶液的浓度优选为3~6mol/l。在本发明中,所述析晶的温度优选为0~30℃,更优选为0~10℃。
31.在本发明中,所述2,4-二氨基-6-氯嘧啶-1-氧化物为淡黄色。在本发明的具体实施例中,所述2,4-二氨基-6-氯嘧啶-1-氧化物的纯度为99.38~99.74%,收率为94.2~98.24%。
32.得到2,4-二氨基-6-氯嘧啶-1-氧化物后,本发明将所述2,4-二氨基-6-氯嘧啶-1-氧化物和哌啶混合,进行氮氧化反应,得到米诺地尔。在本发明中,所述2,4-二氨基-6-氯嘧啶-1-氧化物和哌啶的摩尔比优选为10~20:1,更优选为16~18:1。在本发明中,所述混合优选在搅拌条件下进行。
33.在本发明中,所述氮氧化反应的温度优选为100~106℃,更优选为103~105℃。在本发明中,所述氮氧化反应的时间优选为3h以上,更优选为4~12h。本发明优选采用tlc监测反应终点。
34.在本发明中,所述氮氧化反应后,优选将所得氮氧化反应体系搅拌降温,过夜析晶,抽滤;将所得滤饼用哌啶洗涤,干燥后,得到米诺地尔粗品。在本发明中,所述析晶的温度优选为0~30℃,更优选为0~10℃。在本发明中,所述米诺地尔粗品为类白色固体。
35.在本发明的具体实施例中,所述米诺地尔粗品的纯度为97.39~98.29%,收率为100%。
36.得到米诺地尔粗品后,本发明优选将所述米诺地尔粗品和精制溶剂混合,进行精制,得到精制体系;将所述精制体系依次进行析晶和抽滤,得到滤饼;将所述滤饼依次进行洗涤和干燥,得到米诺地尔。在本发明中,所述精制溶剂优选包括异丙醇、甲醇、乙醇、丙酮和水中的一种或多种,更优选为异丙醇和水的混合液。在本发明中,所述水优选为纯化水。在本发明中,所述异丙醇和水的混合液中异丙醇与水的体积比优选为0.3~0.8:1。在本发明中,所述精制溶剂与米诺地尔粗品的体积比优选为5~8:1。在本发明中,所述精制优选在回流条件下进行,所述精制的温度优选为75~80℃;所述精制的时间优选为1h。在本发明中,所述析晶的温度优选为0~10℃。在本发明中,所述洗涤用溶剂优选为异丙醇。
37.在本发明中,所述米诺地尔为类白色固体。
38.本发明使用的溶剂绿色环保、廉价易得,适合进行工业化生产。在本发明的具体实施例中,所得米诺地尔的纯度为99.81~100.0%,收率为85.1~86.2%。
39.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
40.实施例1
41.a.2,4-二氨基-6-氯嘧啶-1-氧化物的合成
42.向双层玻璃反应釜中加入2.78mol 2,4-二氨基-6-氯嘧啶和6.3kg无水乙醇,搅拌升温至35℃,使体系溶清,搅拌降温至20℃;将3.32mol间氯过氧苯甲酸和2.26kg无水乙醇配制的间氯过氧苯甲酸溶液滴加入上述反应釜中,滴加速度为0.5l/min,滴加完毕,室温反应5小时,tlc色谱板监测反应终点。反应完全后,向体系中加入494g、6mol/l的氢氧化钠溶液;搅拌降温至5℃,过夜析晶,抽滤,滤饼用无水乙醇洗涤一次,干燥,得淡黄色固体2,4-二氨基-6-氯嘧啶-1-氧化物438.5g,产品纯度为99.38%,收率为98.24%。
43.b.米诺地尔粗品的合成
44.向双层玻璃反应釜中加入2.73mol 2,4-二氨基-6-氯嘧啶-1-氧化物和3766.8g哌啶,搅拌升温至103℃,回流反应4小时,tlc色谱板监测反应终点。反应完全后,搅拌降温至5℃,过夜析晶,抽滤,滤饼用哌啶洗涤一次,干燥,得类白色固体米诺地尔粗品580.0g,产品纯度为98.29%,收率为100%。
45.c.米诺地尔的合成
46.向双层玻璃反应釜中加入570.0g所述米诺地尔粗品、1.35kg异丙醇和1.71kg纯化水,搅拌升温至76℃,回流反应1小时,使体系溶清;搅拌降温至5℃,过夜析晶,抽滤,滤饼用异丙醇洗涤一次,干燥,得类白色固体米诺地尔485.1g,产品纯度为99.81%,收率为85.1%。
47.本实施例制备米诺地尔的化学反应式为:
[0048][0049]
实施例2
[0050]
a.2,4-二氨基-6-氯嘧啶-1-氧化物的合成
[0051]
向双层玻璃反应釜中加入13.89mol 2,4-二氨基-6-氯嘧啶和31.6kg无水乙醇,搅拌升温至35℃,使体系溶清,搅拌降温至20℃;将16.63mol间氯过氧苯甲酸和11.3kg无水乙醇配制的间氯过氧苯甲酸溶液滴加入上述反应釜中,滴加速度为0.5l/min,滴加完毕,室温反应5小时,tlc色谱板监测反应终点。反应完全后,向体系中加入2.47kg、6mol/l的氢氧化钠溶液;搅拌降温至5℃,过夜析晶,抽滤,滤饼用无水乙醇洗涤一次,干燥,得淡黄色固体2,4-二氨基-6-氯嘧啶-1-氧化物2.10kg,产品纯度为99.74%,收率为94.2%。
[0052]
b.米诺地尔粗品的合成
[0053]
向双层玻璃反应釜中加入13.08mol 2,4-二氨基-6-氯嘧啶-1-氧化物和18.06kg哌啶,搅拌升温至103℃,回流反应4小时,tlc色谱板监测反应终点。反应完全后,搅拌降温至5℃,过夜析晶,抽滤,滤饼用哌啶洗涤一次,干燥,得类白色固体米诺地尔粗品2.74kg,产品纯度为97.39%,收率为100%。
[0054]
c.米诺地尔的合成
[0055]
向双层玻璃反应釜中加入2.6kg所述米诺地尔粗品、6.1kg异丙醇和7.8kg纯化水,搅拌升温至76℃,回流反应1小时,使体系溶清;搅拌降温至5℃,过夜析晶,抽滤,滤饼用异丙醇洗涤一次,干燥,得类白色固体米诺地尔2.24kg,产品纯度为100%,收率为86.2%。
[0056]
对比例1
[0057]
以中国专利cn107129470a中的实施例2作为对比例,发现采用乙腈作为溶剂时,tlc检测反应不完全或不反应。
[0058]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。