1.本发明属于医药化工领域,具体涉及一种药物中间体的制备方法,特别涉及一种医药中间体6,6-二甲基-3-氮杂双环[3.1.0]己烷或其衍生物的制备方法
背景技术:
[0002]
药物中间体化合物是制药工业中重要的供应链环节,该供应链的产量和质量的稳定性越来越受到整个行业的重视,尤其是在传染性疾病的流行导致药品需求量猛增的时期。
[0003]
已知的是,6,6-二甲基-3-氮杂双环[3.1.0]己烷(6,6-dimethyl-3-azabicyclo[3.1.0]hexane;cas号:943516-54-9)是一种重要的医药中间体,它是很多药物如丙肝蛋白酶抑制剂博赛泼维(boceprevir)和治疗新冠病毒的口服药(pf-07321332)合成过程中所使用的重要原料。
[0004]
它们分子式如下:
[0005][0006]
常见的6,6-二甲基-3-氮杂双环[3.1.0]己烷的合成途径主要是以菊酸乙酯、功夫酸或羟基被保护的异戊烯醇作为原料,先合成中间体卡龙酸酐,卡龙酸酐再经过胺化和还原反应即可制备出6,6-二甲基-3-氮杂双环[3.1.0]己烷。
[0007]
文献1开发了一种以羟基被保护的异戊烯醇和重氮乙酸乙酯为原料制备卡龙酸的方法,其主要步骤如下:
[0008][0009]
该方法以异戊烯醇酯为原料,经过重氮基团的环化反应、水解、氧化得到卡隆酸。但该方法反应步骤长,原子经济性很低,先需使用大量氧化剂将蒈醛酸或蒈醛酸内酯氧化成卡龙酸,不仅顺式产物收率低,而且产生大量废水、废盐,造成严重环境污染。
[0010]
进一步地,文献2公开了一种以卡龙酸酐为起始原料制备6,6-二甲基-3-氮杂双环[3.1.0]己烷的合成路线如下:
[0011][0012]
该方法需要使用大量昂贵的氢化铝锂还原剂将卡龙酸酐还原得到6,6-二甲基-3-氮杂双环[3.1.0]己烷,生产成本高。
[0013]
当然还有其他研究人员对6,6-二甲基-3-氮杂双环[3.1.0]己烷及其前体卡龙酸(酐)的合成路线进行了一定探讨,但考虑到目前以及未来丙肝以及新冠肺炎治疗带来的迅猛需求,如何得到一种绿色环保、原子经济性高的氮杂双环衍生物制备的新方法是目前亟需解决的难题。
[0014]
引用文献:
[0015]
文献1:cn104163759b
[0016]
文献2:cn101384551b
技术实现要素:
[0017]
发明要解决的问题
[0018]
如前所述,现有技术虽然提供了一些6,6-二甲基-3-氮杂双环[3.1.0]己烷及其前体卡龙酸(酐)的制备合成路线,但在长期的工业合成实践中也发现了如下的问题:
[0019]
文献1以异戊烯醇或其衍生物为原料,先制备得到卡隆酸、卡龙酸酐,再由卡龙酸酐还原得到6,6-二甲基-3-氮杂双环[3.1.0]己烷。该方法反应步骤长,原子经济性低。在卡隆酸制备中需要使用大量的氧化剂,产生大量废水和废盐,环境污染严重;所制备得到的卡隆酸是个顺反混合物,需要将反式卡隆酸酐在高温下异构成顺式卡隆酸酐,增加了步骤同时能耗巨大。此外还需还原卡龙酸酐的两个羰基,使价格昂贵的氢化铝锂还原剂用量大幅增加,造成生产成本居高不下。
[0020]
文献2中使用卡龙酸酐作为起始原料,可以便利地得到6,6-二甲基-3-氮杂双环[3.1.0]己烷。但该方法中卡隆酸酐是以菊酸乙酯或功夫酸为原料,这些原料难以制备,生产厂家少规模有限,所合成的卡隆酸酐成本高且无法满足日益增长的用量需求。
[0021]
基于现有技术存在的上述不足,本发明提供了一种新的6,6-二甲基-3-氮杂双环[3.1.0]己烷及其功能性衍生物的合成方法。该方法在配体存在下,利用分子内的环化反应,得到了高顺式选择性的杂原子双环化合物,之后并经由(胺化反应和)还原反应最终到
上述目标产物,革除了氧化步骤从而大大减少了废水废盐的排放;高顺式选择性避免了反式异构体高温异构过程的能耗;绕过了氧化再还原过程,并使还原剂的用量大幅减少。该方法绿色、温和、原子经济性高,适合于工业化大规模生产,为合成下游产品如丙肝蛋白酶抑制剂类药物化合物或治疗新型冠状病毒(covid-19)等药物提供了原料保证。
[0022]
用于解决问题的方案
[0023]
经过本发明人团队长期潜心研究,发现通过如下的技术方案的实施能够解决上述技术问题:
[0024]
[1].本发明首先提供了一种6,6-二甲基-3-氮杂双环[3.1.0]己烷或其衍生物的合成方法,其中,所述方法包括:
[0025]
环化反应的步骤,将通式(1)所表示的化合物进行环化反应以得到通式(2)的化合物所表示的环化产物,
[0026][0027]
其中,
[0028]
所述x表示氧原子或-nh-基团;
[0029]
r表示氢原子或极性基团。
[0030]
[2].根据[1]所述的方法,其中,所述环化反应的步骤,在金属(盐)和有机配体的存在下进行,优选地,所述金属(盐)包括铑、钯、钴、铜及其盐中的一种或多种,所述有机配体包括有机配体包括氮氧、氮氮多齿配体中的一种或多种
[0031]
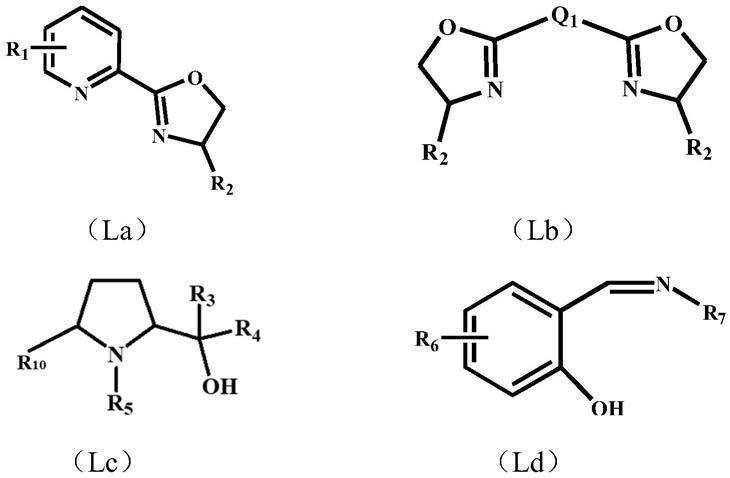
[3].根据[2]所述的方法,其中,所述有机配体选自以下的通式la、lb、lc、ld或le中的一种或多种:
[0032][0033]
式la中,r1可以表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;r2表示氢原子、烷基或芳基;
[0034]
式lb中,q1可以表示二价连接基团或直接键,优选为羰基、具有取代或未取代的亚烷基、具有取代或未取代的芳基亚烷基、具有取代或未取代的亚芳基、具有取代或未取代的亚环烷基、或具有取代或未取代的亚杂芳基;
[0035]
式lc中,r3、r4、r5、r
10
可以相同或不同,表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷氧基、具有取代或未取代的芳氧基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;
[0036]
式ld中,r6、r7可以相同或不同,表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷氧基、具有取代或未取代的芳氧基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;
[0037]
式le中,r8、r9可以相同或不同,表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;q2可以表示二价连接基团或直接键,优选为羰基、具有取代或未
取代的亚烷基、具有取代或未取代的芳基亚烷基、具有取代或未取代的亚芳基、具有取代或未取代的亚环烷基、或具有取代或未取代的亚杂芳基。
[0038]
[4].根据[1]~[3]任一项所述的方法,其中,所述环化反应在有机溶剂中进行;所述环化反应的温度为-10~120℃,优选为60~80℃,反应时间为2~64小时,优选为2~15小时,进一步优选为2~6小时。
[0039]
[5].根据[1]~[4]任一项所述的方法,其中,所述x为氧原子,所述环化产物为通式(2-1)所表示的化合物,进而,所述方法还包括:
[0040]
i)将所述通式(2-1)的化合物进行胺化反应以得到通式(3)的化合物;以及
[0041]
ii)将所述通式(3)的化合物进行还原反应以得到通式(4)的化合物,
[0042][0043]
其中,r的定义与权利要求1中定义相同。
[0044]
[6].根据[1]~[4]任一项所述的方法,其中,所述x为-nh-基团,所述环化产物为通式(3)所表示的化合物,进而,所述方法还包括:
[0045]
将所述通式(3)的化合物进行还原反应以得到通式(4)的化合物,
[0046]
其中,其中通式(3)、通式(4)以及r的定义与权利要求5中的定义相同。
[0047]
[7].根据[1]~[6]任一项所述的方法,其中,所述r为氢原子、羧酸(盐)基、酯基、卤素原子或氰基。
[0048]
[8].进一步,本发明还提供了一种药物中间体的制备方法,其中,所述方法包括根据以上[1]~[7]任一项所述的氮杂双环医药中间体的合成方法。
[0049]
[9].此外,本发明也提供了一种药物化合物的制备方法,其中,所述方法包括根据以上[8]所述的方法。
[0050]
[10].根据[9]所述的方法,其中,所述药物化合物包括丙肝蛋白酶抑制剂类药物化合物或治疗新型冠状病毒(covid-19)的药物化合物。
[0051]
发明的效果
[0052]
通过上述技术方案的实施,本发明能够获得如下的技术效果:
[0053]
1)本发明的制备方法避开制备卡龙酸酐的水解和氧化步骤,大大缩短了制备路线,降低了工业废水、废盐的大量产生,且反应收率高,特别利于工业化大规模生产,可以满足终端药物生产的巨大需求。
[0054]
2)本发明的分子内环化反应催化剂采用铑、钯、钴、铜或铜盐等与配体的组合,收率达98.5%,顺式和反式的比例大于100:1,省略高温异构化步骤,大幅降低生产能耗,同时确保了氮杂双环产物的高产率。
[0055]
3)本发明制备方法还原反应过程中昂贵催化剂的用量大幅度减少,明显提高经济
性。
[0056]
4)本发明提供的合成方法可以灵活地得到多种结构的药物中间体,进而满足不同的药物合成需求。
附图说明
[0057]
图1:本发明一些具体实施方案中合成反应路线图。
具体实施方式
[0058]
以下,针对本发明的内容进行详细说明。以下所记载的技术特征的说明基于本发明的代表性的实施方案、具体例子而进行,但本发明不限定于这些实施方案、具体例子。需要说明的是:
[0059]
本说明书中,使用“数值a~数值b”表示的数值范围是指包含端点数值a、b的范围。
[0060]
本说明书中,使用“以上”或“以下”表示的数值范围是指包含本数的数值范围。
[0061]
本说明书中,使用“可以”表示的含义包括了进行某种处理以及不进行某种处理两方面的含义。
[0062]
本说明书中,使用“任选”或“任选的”表示某些物质、组分、执行步骤、施加条件等因素使用或者不使用以及对使用方式没有限定。
[0063]
本说明书中,所使用的单位名称均为国际标准单位名称,并且如果没有特别声明,所使用的“%”均表示重量或质量百分含量。
[0064]
本说明书中,所提及的“一些具体/优选的实施方案”、“另一些具体/优选的实施方案”、“实施方案”等是指所描述的与该实施方案有关的特定要素(例如,特征、结构、性质和/或特性)包括在此处所述的至少一种实施方案中,并且可存在于其它实施方案中或者可不存在于其它实施方案中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方案中。
[0065]
进一步,本说明书中,以下结构或取代基如无其他特殊说明则按照如下定义解释:
[0066]
术语“烷基”是指脂肪烃基团,其可以是直链或支链的,并且在链中包括1至20个碳原子。优选的烷基在链中包括1至12个碳原子。更优选的烷基在链中包括1至6个碳原子。支链是指一个或多个低级烷基例如甲基、乙基或丙基与直链烷基链相连接。“低级烷基”是指在链中有1至6个碳原子的基团,其可以是直链或支链的。
[0067]
术语“芳基”是指包括6至14个碳原子的芳族单环或多环的环系,优选6至10个碳原子。合适芳基的非限制性例子包括苯基和萘基。
[0068]
术语“芳烷基”是指芳基-烷基-基团,其中芳基和烷基如先前所描述。优选的芳烷基包含低级烷基。合适芳烷基的非限制性例子包括苄基、2-苯乙基和萘基甲基。与母体部分的键是通过烷基连接的。“芳基亚烷基”是指具有芳基取代的亚烷基,优选地,所述芳基亚烷基包括低级亚烷基。并且“低级烷基”是指在链中有1至6个碳原子的亚烷基基团,其可以是直链或支链的。
[0069]
术语“环烷基”是指包括3至10个碳原子的非芳族单或多环系统,优选5至10个碳原子。优选的环烷基环包括5至7个碳原子。合适单环环烷基的非限制性例子包括环丙基,环戊基,环己基,环庚基等等。合适多环环烷基的非限制性例子包括1-十氢化萘,降莰烷基
(norbornyl),金刚烷基等等。
[0070]
术语“卤素原子”是指氟、氯、溴或碘。优选的是氟、氯或溴,且更优选溴和氯。
[0071]
术语“取代的”是指被描述的对象基团可以被一个或多个取代基取代,取代基可以相同或不同,每个取代基独立地选自卤素原子,烷基,芳基,环烷基,氰基,羟基,烷氧基,烷硫基,氨基,-nh(烷基),-nh(环烷基),-n(烷基)2,羧基和-c(o)o-烷基。并且,对于对芳基或杂芳基的取代,作为取代基的芳基可以以并环的方式存在。本段中指出的作为取代基中的“烷基”非限制性例子包括甲基,乙基,正丙基,异丙基,正丁基,叔丁基,正戊基,庚基,壬基,癸基和环丙基甲基。
[0072]
本发明提供了一种6,6-二甲基-3-氮杂双环[3.1.0]己烷或其衍生物的合成方法,通过分子内重氮基团与烯键环化构建杂原子双环化合物,并经由(胺化反应和)还原反应最终得到目标产物。其中,6,6-二甲基-3-氮杂双环[3.1.0]己烷或其衍生物可以作为药物合成中的中间体而被应用。
[0073]
本发明中,所述6,6-二甲基-3-氮杂双环[3.1.0]己烷的衍生物是指下文将述的取代基r的变化,以及取代基r经由化学反应而变化为其他取代基的情况。
[0074]
具体而言,本发明的合成方法首要的包括环化反应的步骤,并且该环化反应实质上是一种重氮基团与烯键的环丙烷化反应。所述环丙烷化反应,通常是与重氮基团相连接的碳与烯键的两个碳进行键联而形成具有环丙烷式三碳环结构。
[0075]
本发明中,所述环化反应将作为起始原料的通式(1)所表示的化合物进行环化反应以得到通式(2)的化合物所表示的环化产物,
[0076][0077]
其中,所述x表示氧原子或-nh-基团。r表示氢原子或极性基团,并且所述极性基团优选选自羧酸(盐)基、酯基、卤原子或氰基。并且,更优选地,所述r为氢原子、甲酸(盐)基或甲酸与烷基、取代的烷基、芳基、取代的芳基、芳烷基或取代的芳烷基、环烷基或取代的环烷基形成的酯基。
[0078]
在本发明一些优选的实施方案中,所述通式(1)的化合物具有如下通式的结构:
[0079][0080]
进一步,对于上述各个结构的化合物,可以通过商购而得到,也可以由实验室合成而得到,例如,可以使用酯部分具有双键的(伯)胺基乙酸酯通过重氮化反应(亚硝酸盐、酸性条件)而得到上述各个化合物。
[0081]
进一步,在一些具体的实施方案中,本发明的环化反应在催化剂以及溶剂的存在下进行;在另外一些具体的实施方案中,本发明的环化反应可以在微波辐照的条件下进行。
[0082]
在一些具体的实施方案中,所述催化剂包括铑、钯、钴、铜等金属(盐)和有机配体。
[0083]
对于金属(盐),其包括金属、金属的无机酸盐、金属的有机酸盐、金属的卤化物中的一种或多种。在本发明一些具体的实施方案中,至少包括铜或铜盐。进一步,本发明的所述铜盐选自氯化亚铜、溴化亚铜、碘化亚铜、三氟甲磺酸亚铜、硫酸铜、醋酸铜、三氟甲磺酸铜以及氯化铜中的一种或多种,优选地,所述铜盐选自氯化亚铜、三氟甲磺酸亚铜、硫酸铜、三氟甲磺酸铜以及氯化铜中的一种或多种。另外,对于金属(盐),除了上述铜(盐)组分以外,也还可以包括其他的金属或金属盐,例如铑、钌等。
[0084]
对于有机配体,本发明中所述有机配体包括氮氧、氮氮多齿配体中的一种或多种。
[0085]
对于氮氧、氮氮多齿配体,其通常通过芳杂环中的氮和或氧实现与金属的配位。所述有机配体选自以下的通式la、lb、lc、ld或le中的一种或多种:
[0086][0087]
式la中,r1可以表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;r2表示氢原子、烷基或芳基;
[0088]
式lb中,q1可以表示二价连接基团或直接键,优选为羰基、具有取代或未取代的亚烷基、具有取代或未取代的芳基亚烷基、具有取代或未取代的亚芳基、具有取代或未取代的亚环烷基、或具有取代或未取代的亚杂芳基;
[0089]
式lc中,r3、r4、r5、r
10
可以相同或不同,表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷氧基、具有取代或未取代的芳氧基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;
[0090]
式ld中,r6、r7可以相同或不同,表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷氧基、具有取代或未取代的芳氧基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具有取代或未取代的环烷基;
[0091]
式le中,r8、r9可以相同或不同,表示氢原子、卤素原子、氰基、硝基、卤代烷基、酯基、具有取代或未取代的烷基、具有取代或未取代的芳烷基、具有取代或未取代的芳基或具
有取代或未取代的环烷基;q2可以表示二价连接基团或直接键,优选为羰基、具有取代或未取代的亚烷基、具有取代或未取代的芳基亚烷基、具有取代或未取代的亚芳基、具有取代或未取代的亚环烷基、或具有取代或未取代的亚杂芳基。
[0092]
在本发明一些具体的实施方案中,对于通式la的配体,可以采用如下结构的有机化合物:
[0093][0094]
对于通式lb的配体,可以采用如下结构的有机化合物:
[0095][0096]
对于通式lc的配体,可以采用如下结构的有机化合物:
[0097][0098]
对于通式ld的配体,可以采用如下结构的有机化合物:
[0099][0100]
对于通式le的配体,可以采用如下结构的有机化合物:
[0101][0102]
进一步,从提高环化产物的收率以及提高环化产物顺式与反式比例的观点考虑,对于优选的有机配体可以为以下l1~l3中的任意一种或多种:
[0103][0104]
对于催化剂中有机配体和金属(盐)的使用比例,在一些具体的实施方案中可以为(摩尔比)1~5:1,优选为2~3:1。另外,对于金属(盐)与式(1)化合物的比例,可以为0.1:1以下,优选为0.01~0.09:1,进一步优选为0.02~0.08:1。
[0105]
进一步,所述环化反应在有机溶剂的存在下进行,对于所述有机溶剂的种类原则上没有特别限制,只要是对于重氮结构的稳定性没有明显影响的有机溶剂即可。在一些优选的实施方案中,所述有机溶剂选自芳香族溶剂、卤化烃类溶剂、砜类溶剂、酰胺类溶剂、乙腈等中的一种或多种的混合溶剂;更优选地,可以选自甲苯(toluene)、二氯乙烷(dce)、二甲基亚砜(dmso)、二甲基甲酰胺(dmf)、乙腈中的一种或多种。
[0106]
对于环化反应条件,在本发明一些具体的实施方案中,环化反应使用溶剂溶解所述催化剂后,在加入式(1)的化合物,优选地,滴加加入式(1)的化合物或者加入溶解了式(1)化合物的溶液,以确保反应的安全性和产率。另外,本发明的环化反应优选的可以在非活性气体的保护下进行,所述非活性气体,可以为氮气、氩气或它们的混合气体。对于反应温度和反应时间,在一些具体的实施方案中,所述环化反应的温度为-10~120℃,优选为60~80℃,反应时间为2~64小时,优选为2~15小时,进一步优选为2~6小时。
[0107]
另外,对于环化反应的其他控制条件,没有特别限制,但优选地,可以在反应的同时进行机械搅拌或磁力搅拌等辅助措施。
[0108]
本发明中,通过上述环化反应,可以得到上述通式(2)结构的化合物。在一些优选的实施方案中,本发明的环化反应的收率可以为75%以上,优选为80%以上,更优选为90%以上。并且,通式(2)结构的化合物的顺式与反式比例(dr值)为大于100:1,具有高的选择性。
[0109]
另外,对于环化反应产物的提纯,典型地,可以通过减压蒸馏等手段对产物进行分离和提纯。
[0110]
得到通式(2)的化合物后,将根据通式(2)的中x原子的具体种类进行后续的合成步骤。
[0111]
在本发明一些具体的实施方案中,通式(2)具有通式(2-1)的结构,
[0112][0113]
进而,本发明所述的合成方法后续还包括:
[0114]
i)将所述通式(2-1)的化合物胺化反应以得到通式(3)的化合物;以及
[0115]
ii)将所述通式(3)的化合物进行还原反应以得到通式(4)的化合物,
[0116][0117]
其中,r的定义与通式(1)中定义相同。
[0118]
在步骤i中,对于胺化反应的具体条件,没有特别限定,可以采用本领域常规的胺化反应条件。
[0119]
在本发明一些具体的实施方案中,所述胺化反应借助氨气的醇类溶剂进行,并且对于醇类溶剂,从便利的角度考虑,优选为甲醇。
[0120]
典型地,可以将通式(2-1)的化合物溶解于醇类溶剂中,并加入氨-醇试剂,并在不高于10℃,优选为-10~5℃,更优选为-5~0℃的条件下进行反应,反应时间没有特别限制,可以为1~3小时。
[0121]
进一步,步骤i反应结束后,可以通过减压蒸馏的手段分离出通式(3)结构的产物。
[0122]
在步骤ii中,将通式(3)的羰基结构进行还原。对于还原反应,可以在借助溶剂和还原剂的条件下进行。对于溶剂,可以使用非质子类溶剂,例如酯类溶剂、酰胺类溶剂、醚类溶剂、环醚类溶剂、酮类溶剂、砜类溶剂、芳香烃类溶剂等中的一种或多种;优选地,可以使用四氢呋喃、1,4-二氧六环、乙醚、苯、甲苯、乙二醇二甲醚中的一种或多种。对于还原剂,选自氢化铝锂、硼氢化钠/三氟化硼乙醚、硼烷、2-氢双(二甲氧乙氧基)铝酸钠中的至少一种,进一步优选地,还原剂为氢化铝锂或硼氢化钠/三氟化硼乙醚。在本发明一些具体的实施方案中,可以使用氢化锂铝作为催化剂。对于步骤ii的其他反应条件,没有特别限制,可以采用本领域常用的还原反应的条件。典型的方式中,例如,还原反应中氢化铝锂与通式(3)化合物的摩尔比为1~4:1,反应时间为2~3小时,反应温度为-5~10℃。待反应结束后,可以通过分相萃取、减压蒸馏而得到通式(4)表示最终的产物。
[0123]
另外,对于本发明的步骤ii,尤其地适合r为氢方案,对于r为极性基团的情况,在任意的必要时,可以使用任选的保护基团对r进行临时保护或者,也可以采用另外的还原方
式对羰基进行还原处理。
[0124]
在一些具体的实施方案中,步骤i和ii的总收率可以为90%以上。
[0125]
在本发明另外一些具体的实施方案中,作为环化产物的通式(2)具有通式(3)的结构,则在环化反应之后的后续反应中,可以直接进行上述步骤ii,以得到最终的通式(4)表示最终的产物。
[0126]
进一步,以附图1对本发明的典型的合成路线做出说明:
[0127]
包括以重氮乙酸(2-甲基-2-丁烯)酯为原料,先分子内环化反应制中间体式a,中间体式a经胺化、还原反应得到产物6,6-二甲基-3-氮杂双环[3.1.0]己烷;
[0128]
或者,以重氮(2-甲基-2-丁烯)乙酰胺为原料,先分子内环化反应制中间体b,中间体式b经一步酰化还原反应得到产物6,6-二甲基-3-氮杂双环[3.1.0]己烷。
[0129]
进一步,通过本发明作为原料的通式(1)中r的取代基的调整,本发明可以得到不同的药物中间体,典型地:
[0130]
当r为氢时,可以得到以下式(a)的化合物;当r为酯基时,可以得到以下式(b)的化合物。并且,不言而喻的,当变化r为其他的极性基团时(例如-cn,如式(c)化合物)。还可以通过后续任意的化学过程将r改变为所要的其他的基团r1(r1可以为药学活性基团),其变化的种类和方式没有特别限制,例如也可以通过式c的化合物中-cn基团的后续处理而得到式b的化合物。
[0131][0132]
进一步,基于本发明的上述所提供的新的合成方法,可以高效率的提供相关药物中间体的生产,可以作为药物合成中的中间合成步骤。
[0133]
同时,本发明的合成方法,也可以提供改进的环保型和合成灵活性,特别适用于稳定、高产量的提供药物合成工业的原料供应。
[0134]
此外,通过本发明合成方法所提供的药物中间体,可以用于包括丙肝蛋白酶抑制剂类药物化合物或治疗新型冠状病毒(covid-19)的药物化合物的制备。
[0135]
实施例
[0136]
以下,将通过具体的实施例对本发明做出进一步的说明。
[0137]
(原料来源)
[0138]
化合物重氮乙酸(2-甲基-2-丁烯)酯(自制得到):
[0139][0140]
化合物重氮(2-甲基-2-丁烯)乙酰胺(自制得到):
[0141][0142]
(中间体式a立体构型):
[0143]
顺式结构
[0144]
反式结构
[0145]
(中间体式b立体构型):
[0146]
顺式结构
[0147]
反式结构
[0148]
实施例1(制备中间体式a)
[0149]
向200ml烧瓶中加入196mg氯化亚铜,加入680mg配体l1,加入二氯乙烷溶液100ml,于室温下搅拌1h,升温至75℃,缓慢滴加15.4g
[0150][0151]
化合物3h,反应结束后减压蒸出产物中间体式a,收率为93.1%,顺式和反式的比例大于100:1。
[0152]
实施例2~14(制备中间体式a)
[0153]
与实施例1区别在于反应参数控制不同,具体反应参数及反应效果如表1所示。
[0154]
实施例15
[0155]
将实施例1中制备得到的12.6g中间体式a溶于甲醇中,加入5mol/l的氨甲醇溶液100ml,于0℃下搅拌1h,生成中间体式b,溶液直接减压蒸馏,得粗品12g。向粗品中加入四氢呋喃溶液150ml0℃下加入氢化铝锂7.6g,搅拌2h,反应结束后加入乙酸乙酯200ml淬灭反应,加入水50ml,萃取分层并合并有机相,有机相通过减压蒸馏获得产品式a化合物10.1g,两步总收率91.0%。
[0156]
实施例16
[0157]
向200ml烧瓶中加入196mg氯化亚铜,加入680mg配体l1,加入二氯乙烷溶液100ml,于室温下搅拌1h,升温至75℃,缓慢滴加15.2g
[0158][0159]
化合物3h,反应结束后减压蒸出中间体式b,产品收率为92.8%,顺式和反式的比例大于100:1。
[0160]
将上述制备得到的12.5g中间体式b加入四氢呋喃溶液150ml;0℃下加入氢化铝锂7.6g,搅拌2h,反应结束后加入乙酸乙酯200ml淬灭反应,加入水50ml,萃取分层并合并有机相,有机相通过减压蒸馏获得式a化合物10.3g,收率92.7%。
[0161]
表1(实施例1~14)反应参数
[0162][0163]
需要说明的是,尽管以具体实例介绍了本发明的技术方案,但本领域技术人员能够理解,本公开应不限于此。
[0164]
以上已经描述了本公开的各实施例,上述说明是示例性的,并非穷尽性的,并且也不限于所披露的各实施例。在不偏离所说明的各实施例的范围和精神的情况下,对于本技术领域的普通技术人员来说许多修改和变更都是显而易见的。本文中所用术语的选择,旨在最好地解释各实施例的原理、实际应用或对市场中的技术的改进,或者使本技术领域的其它普通技术人员能理解本文披露的各实施例。
[0165]
产业上的可利用性
[0166]
本发明提供的合成方法可以在工业上制备医药中间体化合物。