1.本发明高分子复合材料技术领域,具体指一种高强高韧阻燃型聚乳酸复合材料及其制备方法。
背景技术:
2.聚乳酸是一种100%来自可再生原料,生物可降解且生物相容性好的热塑性塑料,其加工性能优良,具有广阔的工业化前景,是近些年来开发研究最活跃、发展最快的聚烯烃树脂替代品。随着pla应用范围的进一步拓展,对其性能也有了更高的要求。聚乳酸材料本身具有较高的抗张强度和拉伸模量,但是由于聚乳酸分子链缠结密度低且特征比高,其冲击强度极低且延展性极小。此外,近年来聚乳酸在电器、办公室自动化设备的壳体等领域得到了越来越多的应用,防火性能显得更加重要。因此,制备高抗张强度、高延展性、高冲击强度、阻燃型聚乳酸材料具有重要的意义。
3.近来,无卤型阻燃剂的研究越来越受到重视。有机磷化合物是一种重要的无卤型阻燃剂。比如,磷酸酯齐聚物已经被应用于多种树脂材料的阻燃。此外,磷酸酯齐聚物是一种柔性聚合物,将其作为添加剂与其他高分子树脂材料共混的方法可有效改善脆性树脂材料的延展性和冲击强度。但是此类磷酸酯齐聚物稳定性相对较差,且与聚乳酸树脂基体的相容性较差,易析出。如何改善聚磷酸酯与聚乳酸基体的相容性缺陷是拓展其应用价值的重要工作。
4.专利申请号为cn201610870221.3(公布号为cn106433055a)的发明专利《一种具有良好阻燃性的高强度生物质板材的制备方法》公开了一种阻燃性聚乳酸树脂组合物,其在sncl2的催化下,通过扩链剂二氯膦酸乙酯与聚乳酸的端羟基在熔融共混状态下的原位反应,将磷元素引入聚乳酸聚合物链中实现阻燃性能。
5.但上述方案在熔融共混过程中进行原位反应,将磷元素引入聚乳酸聚合物链中实现阻燃性能,操作复杂,成本较高,每次制备复合材料都需要反复进行共聚反应,同时还存在一些问题:(1)二氯膦酸乙酯与羟基的反应会释放有毒的氯化氢,对环境有害,且严重危害操作人员的健康;(2)锡为具有较高毒性的重金属元素,残留的锡元素会严重损害使用者的健康;(3)通过该方法制备的聚乳酸板材,拉伸强度只有约40mpa,大大低于纯聚乳酸材料。
技术实现要素:
6.本发明所要解决的第一个技术问题是针对现有技术的现状,提供一种制备简单、综合力学性能较普通聚乳酸材料有较大提升,且具有阻燃特性的高强高韧阻燃型聚乳酸复合材料。
7.本发明所要解决的第二个技术问题是提供一种上述高强高韧阻燃型聚乳酸复合材料的制备方法,该制备方法操作简单。
8.本发明解决第一个技术问题所采用的技术方案为:一种高强高韧阻燃型聚乳酸复
合材料,其特征在于,按质量份数计,包括有以下组分:
9.聚乳酸40~99份,
10.多嵌段共聚物1~60份;
11.所述的多嵌段共聚物至少包含一个聚乳酸链段和一个聚磷酸酯链段,结构式如下:
[0012][0013]
其中,r1基团为氢、c1~c20的烷基、c1~c20的芳基、c1~c20的烷氧基或c1~c20的芳氧基;
[0014]
r2基团为c1-c20的烷基、c1-c20的芳基、c1-c20的烷氧基、c1-c20的烯基或c1-c20的炔基;
[0015]
n、x、y均为正整数;
[0016]
所述聚磷酸酯链段的含量为1~75wt%;
[0017]
所述多嵌段共聚物的重均分子量≥1万。
[0018]
优选地,所述多嵌段共聚物的合成方法包括有以下步骤:按质量份数,在无水、无氧的条件下,将0.01~5份引发剂、0.01~5份催化剂与溶剂混合形成溶液,将20~99份丙交酯单体加入到聚合体系中进行聚合,聚合温度为-40℃~60℃,聚合时间为10s~1h;加入1~80份环磷酸酯单体进行聚合,聚合温度为-40℃~60℃,聚合时间为30s~2h;加入0.01~5份化合物a进行反应,反应温度为-40℃~60℃,反应时间为30s~5h,得到所需的多嵌段共聚物;
[0019]
所述化合物a的分子结构中有且仅有两个异氰酸酯基团。
[0020]
其中,化合物a作为扩链剂参与反应,其作用是将原先分子量较小的嵌段共聚物进行扩链反应,使其转化为具有更高分子量的多嵌段共聚物,目的是避免较小分子量添加剂的加入导致的力学性能大幅下降。
[0021]
进一步,所述的引发剂为二醇类化合物。
[0022]
更进一步,所述的二醇化合物为乙二醇、对苯二甲醇、1,4-丁二醇、1,6-己二醇中的至少一种。
[0023]
进一步,所述的催化剂为有机碱。与传统金属催化剂相比,聚合条件更为温和,价格更为低廉,同时还避免了有毒重金属的残留。
[0024]
更进一步,所述的5为1,8-二氮杂二环[5.4.0]十一碳-7-烯、1,5,7-三氮杂二环[4.4.0]癸-5-烯、吡啶、4-二甲氨基吡啶、鹰爪豆碱中的至少一种。
[0025]
进一步,所述溶剂为非质子性溶剂。
[0026]
更进一步,所述的溶剂为乙酸乙酯、n,n-二甲基甲酰胺、二甲亚砜、氯仿、二氯甲烷、四氢呋喃中的至少一种。
[0027]
进一步,所述的丙交酯单体为左旋丙交酯、右旋丙交酯、内消旋丙交酯中的至少一种。
[0028]
进一步,所述环磷酸酯单体的结构式如下:
[0029][0030]
其中,r1基团为氢、c1~c20的烷基、c1~c20的芳基、c1~c20的烷氧基或c1~c20的芳氧基;
[0031]
r2基团为c1-c20的烷基、c1-c20的芳基、c1-c20的烷氧基、c1-c20的烯基或c1-c20的炔基。
[0032]
进一步,所述的化合物a为1,6-二异氰酸酯基己烷、二甲苯烷二异氰酸酯、三甲基六亚甲基二异氰酸酯、甲苯-2,4-二异氰酸酯、异氟尔酮二异氰酸酯中的至少一种。
[0033]
优选地,所述的聚乳酸链段为左旋聚乳酸、右旋聚乳酸、左旋右旋乳酸无规排列的无规聚乳酸中的至少一种。
[0034]
优选地,所述聚磷酸酯链段的含量为1~60wt%,所述多嵌段共聚物的重均分子量≥3万。
[0035]
本发明解决第二个技术问题所采用的技术方案为:一种上述高强高韧阻燃型聚乳酸复合材料的制备方法,其特征在于包括以下步骤:将商业化聚乳酸粒料和多嵌段共聚物在160℃~200℃的条件下熔融共混制得所需的高强高韧阻燃型聚乳酸复合材料。
[0036]
与现有技术相比,本发明的优点在于:
[0037]
(1)本发明使用含有聚乳酸和聚磷酸酯链段的多嵌段共聚物链段作为添加剂,其中,聚磷酸酯是一种柔性的阻燃的聚合物,向复合材料中添加聚磷酸酯可以提高聚合物的延展性和抗冲击强度,并赋予复合材料阻燃性能;而聚乳酸链段使多嵌段共聚物与聚乳酸基体材料的相容性增加,防止聚磷酸酯在复合材料中析出,从而削弱复合材料的性能;
[0038]
(2)本发明先通过聚乳酸和聚磷酸酯共聚形成多嵌段共聚物,然后将该多嵌段共聚物以添加剂的形式添加到聚乳酸基体中,制备简单,储存方便,只需要进行一次共聚反应,无需在下游的复合材料共混过程中反复通过共聚反应引入聚磷酸酯链段;
[0039]
(3)本发明采用丙交酯与环磷酸酯开环共聚的方法制备嵌段共聚物,具有条件温和、聚合效果好的优点;此外,环磷酸酯开环聚合的方法温和地、环境友好地引入了聚磷酸酯链段,避免了磷酰氯等的使用,减少了有毒、污染性气体的产生;
[0040]
(4)本发明的聚乳酸复合材料具有抗张强度高、延展性好、抗冲击强度高、阻燃效率高、无卤、无腐蚀性、环境友好等优点,同时,拉伸强度最高可达53.1mpa,虽与纯pla相比略有下降,但是,断裂伸长率最高可达221.7%,与纯pla相比提高了约30倍,冲击强度与纯pla相比也有18%~38%的提高,可广泛用于电器通讯组件和包装材料的制造。
附图说明
[0041]
图1为本发明实施例所制备的多嵌段共聚物a和b的凝胶渗透色谱图;
[0042]
图2为本发明实施例1~2所制备的多嵌段共聚物a的核磁共振氢谱图;其中5.1-5.2ppm的多重峰和1.5-1.7ppm的双重峰为聚乳酸链段的共振峰(其中5.1-5.2ppm的多重峰的积分面积记为m1),化学位移为3.8ppm的三重峰和4.2-4.5ppm处的多重峰为聚磷酸酯链段的共振峰(积分面积为m2);所得多嵌段共聚物中聚磷酸酯链段的含量可以用以下公式计算:(m1
÷2×
144.14)
÷
(m1
÷2×
144.14+m2
÷2×
138.06)=9.3%,其中,144.14为聚乳酸
结构单元分子量,138.06是聚磷酸酯结构单元的分子量;
[0043]
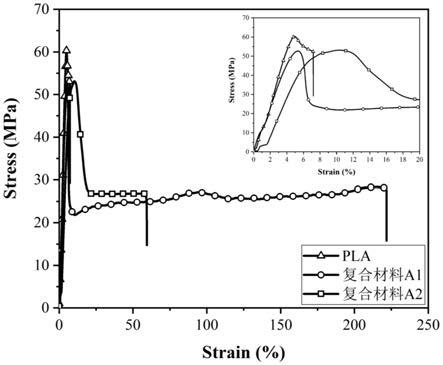
图3为本发明实施例1~2所制备的复合材料a1和a2的应力应变曲线。
具体实施方式
[0044]
以下结合附图实施例对本发明作进一步详细描述。
[0045]
比较例1:
[0046]
将40g的pla纯料(本发明比较例和实施例所用的pla原料均为natureworks,no.4032d)在190℃的挤出机进行挤出造粒,最后烘干得到粒料。
[0047]
性能测试:
[0048]
①
阻燃测试评价:
[0049]
上述粒料经充分干燥后,在200℃的条件下注塑成型,从而制备阻燃性能评价的样条(125mm*13mm*1.6mm);以ul(underwriters laboratories)-94(标准见表1)为基准,对该样条实施了20mm垂直燃烧实验(燃烧测试结果见2);根据实验结果,该样品在ul-94的标准下没有等级;
[0050]
②
力学性能评价:
[0051]
将上述粒料在190℃下进行注塑,得到拉伸国标样条以及自动生成缺口的国标冲击样条,随后采用5569instron万能拉伸测试机并按照国标gb/t1040.2-2006测试其拉伸性能,采用jxud5.5摆锤冲击测试仪并按照国标gb/t1843-2008测试其冲击强度,拉伸强度为60.3mpa,断裂伸长率为7.2%,冲击强度为3.2kj/m2。
[0052]
实施例1:
[0053]
(1)多嵌段共聚物a的合成:
[0054]
在无水、无氧的条件下,量取0.1ml乙二醇(1.8mmol)作为引发剂,称取0.137gdbu(0.9mmol)作为催化剂,与20ml二氯甲烷混合形成溶液,将25.92g左旋丙交酯单体(180mmol)加入到聚合体系中,聚合温度保持在25℃,聚合时间40min;加入2-甲氧基-1,3,2-二氧磷杂环戊烷-2-氧化物(环磷酸酯单体a,式3)6.21g(45mmol),聚合温度25℃,聚合时间20min;加入1,6-二异氰酸酯基己烷0.303g(1.8mmol),反应温度25℃,反应时间2h;聚合结束后,用200ml乙醚沉降出聚合物,在50℃真空烘箱中抽干至恒重,得到多嵌段共聚物a;采用凝胶渗透色谱和核磁共振氢谱的方法对多嵌段共聚物a进行表征(凝胶渗透色谱图见图1,核磁共振氢谱图见图2),所得聚合物的重均分子量mw为122.8kg/mol,分子量分布1.55,聚磷酸酯的含量为9.3wt%;
[0055][0056]
(2)复合材料a1的制备:
[0057]
将20g的pla纯料与20g多嵌段共聚物1加入到高速共混机(转速300rpm/min),初混5min后得到预混物,再将其在190℃的挤出机进行挤出造粒,最后烘干得到复合材料a1的粒料。
[0058]
性能测试:
[0059]
①
阻燃测试评价:
[0060]
复合材料a1的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备垂直燃烧评价的样条(125mm*13mm*1.6mm);以ul(underwriters laboratories)-94(标准见表1)为基准,对该样条实施了20mm垂直燃烧实验(燃烧测试结果见2);根据实验结果,该样品被评价为ul-94标准的v-0;
[0061]
复合材料a1的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备极限氧指数(loi)评价的样条(100mm*6.5mm*3mm);用氧指数仪对其进行测试,loi值为32.1
±
0.2vol%;
[0062]
②
力学性能评价:
[0063]
将复合材料a1的粒料在190℃下进行注塑,得到拉伸国标样条以及自动生成缺口的国标冲击样条,随后采用5569instron万能拉伸测试机并按照国标gb/t1040.2-2006测试其拉伸性能,采用jxud5.5摆锤冲击测试仪并按照国标gb/t1843-2008测试其冲击强度,拉伸强度为52.7mpa,与纯pla相比拉伸强度只降低了12.6%;断裂伸长率为221.7%,与纯pla相比增加了近30倍;冲击强度为4.4kj/m2,与纯pla相比增大了37.5%,其应力应变曲线见图3。
[0064]
实施例2:
[0065]
(1)多嵌段共聚物a的合成:同实施例1;
[0066]
(2)复合材料a2的制备:
[0067]
将28g的pla纯料与12g多嵌段共聚物a加入到高速共混机(转速300rpm/min),初混5min后得到预混物,再将其在190℃的挤出机进行挤出造粒,最后烘干得到复合材料a2的粒料。
[0068]
性能测试:
[0069]
①
阻燃测试评价:
[0070]
复合材料a2的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备阻燃性能评价的样条(125mm*13mm*1.6mm);以ul-94为基准,对该样条实施了20mm垂直燃烧实验(燃烧测试结果见2);根据实验结果,该样品被评价为ul-94标准的v-0;
[0071]
复合材料a2的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备极限氧指数(loi)评价的样条(100mm*6.5mm*3mm);用氧指数仪对其进行测试,loi值为30.9
±
0.1vol%;
[0072]
②
力学性能评价:
[0073]
将复合材料a2的粒料在190℃下进行注塑,得到拉伸国标样条以及自动生成缺口的国标冲击样条,随后采用5569instron万能拉伸测试机并按照国标gb/t1040.2-2006测试其拉伸性能,采用jxud5.5摆锤冲击测试仪并按照国标gb/t1843-2008测试其冲击强度,拉伸强度为53.1mpa,与纯pla相比拉伸强度只降低了11.9%;断裂伸长率为59.2%,与纯pla相比增加了7.2倍;冲击强度为3.9kj/m2,与纯pla相比增大了21.9%,其应力应变曲线见图3。
[0074]
实施例3:
[0075]
(1)多嵌段共聚物b的合成:
[0076]
在无水、无氧的条件下,量取0.1ml乙二醇(1.8mmol)作为引发剂,称取0.137gdbu(0.9mmol)作为催化剂,与20ml二氯甲烷混合形成溶液,将25.92g左旋丙交酯单体
(180mmol)加入到聚合体系中,聚合温度保持在25℃,聚合时间40min;加入2-乙氧基-1,3,2-二氧磷杂环己烷-2-氧化物(环磷酸酯单体b,式4)7.47g(45mmol),聚合温度25℃,聚合时间20min;加入1,6-二异氰酸酯基己烷0.303g(1.8mmol),反应温度25℃,反应时间2h;聚合结束后,用200ml乙醚沉降出聚合物,在50℃真空烘箱中抽干至恒重,得到多嵌段共聚物b;采用凝胶渗透色谱和核磁共振氢谱的方法对多嵌段共聚物b进行表征(凝胶渗透色谱图见图1),所得聚合物的重均分子量mw为165.5kg/mol,分子量分布1.71,聚磷酸酯的含量为9.1wt%;
[0077][0078]
(2)复合材料b1的制备:
[0079]
将20g的pla纯料与20g多嵌段共聚物b加入到高速共混机(转速300rpm/min),初混5min后得到预混物,再将其在190℃的挤出机进行挤出造粒,最后烘干得到复合材料b1的粒料。
[0080]
性能测试:
[0081]
①
阻燃测试评价:
[0082]
复合材料b1的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备阻燃性能评价的样条(125mm*13mm*1.6mm);以ul-94为基准,对该样条实施了20mm垂直燃烧实验(燃烧测试结果见2);根据实验结果,该样品被评价为ul-94标准的v-0;
[0083]
复合材料b1的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备极限氧指数(loi)评价的样条(100mm*6.5mm*3mm);用氧指数仪对其进行测试,loi值为31.9
±
0.1vol%;
[0084]
②
力学性能评价:
[0085]
将复合材料b1的粒料在190℃下进行注塑,得到拉伸国标样条以及自动生成缺口的国标冲击样条,随后采用5569instron万能拉伸测试机并按照国标gb/t1040.2-2006测试其拉伸性能,采用jxud5.5摆锤冲击测试仪并按照国标gb/t1843-2008测试其冲击强度,拉伸强度为52.9mpa,与纯pla相比拉伸强度只降低了12.3%;断裂伸长率为214.5%,与纯pla相比增加了约29倍;冲击强度为4.1kj/m2,与纯pla相比增大了28.1%。
[0086]
实施例4:
[0087]
(1)多嵌段共聚物b的合成:同实施例3;
[0088]
(2)复合材料b2的制备:
[0089]
将28g的pla纯料与12g多嵌段共聚物b加入到高速共混机(转速300rpm/min),初混5min后得到预混物,再将其在190℃的挤出机进行挤出造粒,最后烘干得到复合材料b2的粒料。
[0090]
性能测试:
[0091]
①
阻燃测试评价:
[0092]
复合材料b2的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备阻燃性能评价的样条(125mm*13mm*1.6mm);以ul-94为基准,对该样条实施了20mm垂直燃烧实验(燃
烧测试结果见2);根据实验结果,该样品被评价为ul-94标准的v-0;
[0093]
复合材料b2的粒料经充分干燥后,在200℃的条件下注塑成型,从而制备极限氧指数(loi)评价的样条(100mm*6.5mm*3mm);用氧指数仪对其进行测试,loi值为30.6
±
0.2vol%;
[0094]
②
力学性能评价:
[0095]
将复合材料b2的粒料在190℃下进行注塑,得到拉伸国标样条以及自动生成缺口的国标冲击样条,随后采用5569instron万能拉伸测试机并按照国标gb/t1040.2-2006测试其拉伸性能,采用jxud5.5摆锤冲击测试仪并按照国标gb/t1843-2008测试其冲击强度,拉伸强度为51.3mpa,与纯pla相比拉伸强度只降低了14.9%;断裂伸长率为69.7%,与纯pla相比增加了约8.7倍;冲击强度为3.8kj/m2,与纯pla相比增大了18.8%。
[0096]
表1阻燃测试ul-94标准
[0097]
试样燃烧行为v-0v-1v-2每个试样每次点燃后单个试样最长有焰燃烧时间(t1或t2)≤10s≤30s≤30s第二次点燃后单个试样最长无焰燃烧时间(t2+t3)≤30s≤60s≤60s点燃后最长有焰燃烧总时间(t1+t2)≤50s≤250s≤250s有无熔滴和熔滴是否引燃棉花否否是是否烧到固定夹否否否
[0098]
表2试样燃烧测试结果
[0099]