1.本发明属于碳纳米材料制备和发光材料制备技术领域,具体涉及一种碳纳米点/硼酸复合磷光材料及其制备方法和应用。
背景技术:
2.在实际应用中,高性能的光电器件不仅需要高的室温磷光(rtp)发射效率,同时还需要高的rtp发射强度,所以兼具高效率和高强度的室温磷光材料在防伪、传感器和光电子器件等广泛应用中具有重要意义。到目前为止,高效rtp材料仍大多限于金属掺杂无机配合物和有机金属化合物等。但是金属掺杂无机配合物材料中含有贵金属,通常存在成本高、毒性大等缺点,不适合实际应用。而有机金属化合物光稳定性较差、制备过程繁琐、复杂,难以实际应用。
3.碳纳米点是近年来发展来的一种碳纳米磷光材料,与传统rtp磷光材料相比,具有制作成本低、发光性能稳定、易于功能化和无毒等优势,在光电器件、数字加密和防伪等领域具有广阔的应用前景。然而,现有的碳纳米点材料磷光发射效率相仍较低,通常小于20%,且发射强度较低,到目前为止仍缺乏一种兼具高效率和高强度磷光碳纳米点复合材料。
技术实现要素:
4.针对现有技术中存在不足,本发明提供了一种碳纳米点/硼酸复合磷光材料及其制备方法和应用。在本发明中,首先将碳纳米点和硼酸基质混合,通过高温长时间熔融处理,使碳纳米点充分、最大程度的分散到硼酸基质中,然后通过研磨诱导硼酸基质发生无定形-结晶晶型转变,从而原位的将碳纳米点镶嵌在晶体硼酸基质中得到所述碳纳米点/硼酸复合磷光材料;所述碳纳米点/硼酸复合磷光材料具有高效、高强度的磷光发射,且制备方法简单,成本低廉。
5.本发明中首先提供了一种碳纳米点/硼酸复合磷光材料,所述碳纳米点/硼酸复合磷光材料中碳纳米点原位镶嵌在硼酸晶体中;所述复合磷光材料的粒径为5~200 μm,所述复合磷光材料在紫外灯光关闭后可发出明亮的黄绿色磷光。
6.本发明中还提供了上述碳纳米点/硼酸复合磷光材料的制备方法,具体包括如下步骤:将碳纳米点和硼酸基质在去离子中混合均匀,然后在150~350℃下加热反应,反应结束后冷却、研磨,得到所述碳纳米点/硼酸复合磷光材料。
7.进一步的,所述碳纳米点、硼酸基质和去离子水的质量比为1:100~500:800~6000。
8.进一步的,所述加水热反应温度为200℃。
9.进一步的,所述加热反应的时间为1-8h。
10.进一步的,所述加热反应的时间为5h。
11.本发明中还提供了上述碳纳米点/硼酸复合磷光材料在制备光电器件中的应用。
12.本发明中还提供了上述碳纳米点/硼酸复合磷光材料在数字加密和防伪领域中的
应用。
13.与现有技术相比,本发明的有益效果在于:相比于传统的结晶方法,本发明的提出的研磨诱导无定形-晶体转变方法可使得碳纳米点最大程度的、无明显聚集的分散到硼酸晶体中,极大提高了碳纳米点在晶体中的负载量。同时基质强刚性晶体环境,极大抑制了碳点非辐射跃迁和促进系间窜越效率,从而获得高效、高强度的磷光发射。
14.本发明中制备的碳纳米点硼酸复合材料,磷光发光效率高达48%,是目前碳纳米点磷光材料的最高纪录,同时复合材料磷光强度在关闭紫外光源9s内可清晰照亮物品,在延迟1s时刻的磷光强度高达590 lux,远高于市面手机、电脑屏幕的光照值。
15.本发明中制备的碳纳米点硼酸复合材料,可被太阳光激发产生明亮的黄绿色磷光,即使阴天或者雨天太阳光的环境下,材料仍可发出明亮的磷光,人眼可见寿命长达9s。
附图说明
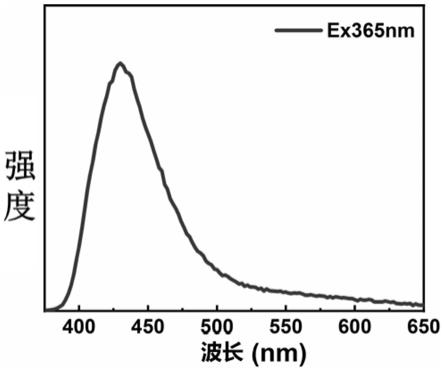
16.图1是碳纳米点溶液在365nm激发下的荧光光谱图。
17.图2是cd@ba粉末365nm激发下的磷光光谱图。
18.图3是cd@ba透的射电镜(tem)图像。
19.图4是cd@ba的扫描电镜(sem)图像。
20.图5是cd@ba的ftir光谱图。
21.图6是cd@ba高分辨率c 1s xps谱图。
22.图7是cd@ba高分辨率b 1s xps谱图。
23.图8是碳纳米点溶液的紫外-可见吸收光谱和原始样品的rtp激发光谱图。
24.图9是cd@ba在阴天阳光和365nm光源激发前后的数码照片。
25.图10是cd@ba研磨前后的x射线衍射(xrd)光谱图。
26.图11是cd@ba研磨前后的差示扫描量热(dsc)曲线图。
27.图12是e-cd@ba和cd@ba不同负载浓度的碳纳米点溶液制备出的样品的数码照片。
28.图13是不同负载浓度的碳纳米点溶液下e-cd@ba和cd@ba的磷光强度图。
具体实施方式
29.下面结合附图以及具体实施例对本发明作进一步的说明,但本发明的保护范围并不限于此。
30.实施例1:碳纳米点溶液的制备称取盐酸石蒜碱0.01g和30ml去离子水放入反应釜中充分混合,再将其置于200℃的恒温鼓风箱中加热2h,取出后待其自然冷却,将反应釜中的液体倒出,得到碳纳米点溶液。
31.图1是碳纳米点溶液在365nm激发下的荧光光谱,图中表明在紫外光激发下的发射峰值在440nm左右。
32.实施例2:碳纳米点/硼酸复合磷光材料的制备取碳纳米点溶液5ml(3.4mg/ml)、硼酸ba 3g和25ml去离子水放入烧杯中充分混合,再将烧杯置于200℃的恒温鼓风箱中加热5h,加热结束后取出使其快速冷却成块,将烧
杯中的块状物取出倒入玛瑙研钵中进行研磨,得到碳纳米点/硼酸复合磷光材料,记为cd@ba,经检测,粉末磷光量子产率为48%。
33.图2是cd@ba粉末365nm激发下的磷光光谱,图中表明在365 nm的紫外光激发下,cd@ba粉末和原始块状体进行比较,研磨后的磷光强度明显高于原始块状体3-4倍,这是因为研磨抑制了碳纳米点的自由振动,减少了非辐射跃迁,促进了隙间跨越,从而产生了高磷光量子产率为48%的强rtp材料。
34.图3是cds透射电镜(tem)图像,从图中可以看出,cds的平均直径为5
ꢀ±ꢀ
1.2 nm。
35.图4是cd@ba的扫描电镜(sem)图像,从图中可以看出,cd@ba的粒径分布为5~200 μm。
36.实施例3:碳纳米点/硼酸复合磷光材料的制备取碳纳米点溶液10ml(3.4mg/ml)、硼酸ba 3g和20ml去离子水放入烧杯中充分混合后于200℃的恒温鼓风箱中加热5h,加热结束后取出使其快速冷却成块,将烧杯中的块状物取出倒入玛瑙研钵中进行研磨,得到的cd@ba粉末磷光量子产率为42%。
37.图5是cd@ba的傅里叶变换红外光谱(ftir)光谱,在图中cds在3400、1631和1617 cm
−1处显示出特征吸收峰,分配给的拉伸振动o
−
h、c=o和c=c。将cds掺入ba后,在941cm
−1处出现新的吸收峰对应于c-b的拉伸振动。
38.图6、7是cd@ba的xps全光谱与高分辨率c 1s 和b 1s xps谱,在图6的xps结果中,有三个以288ev、286.6ev、285.1 ev为中心的峰值,分别属于c=o、c-o/c-n和c-c/c=c。在图7中可看到在以194.6 ev、193.6 ev和192.7 ev的峰值,分别属于b-o、b2o3和bco2。这些结果表明cds成功地嵌入到ba基体中。
39.实施例4:碳纳米点/硼酸复合磷光材料的制备取碳纳米点溶液3ml(3.4mg/ml)、硼酸ba 3g和27ml去离子水放入烧杯中充分混合后于200℃的恒温鼓风箱中加热3h,取出后使其快速冷却成块,将烧杯中的块状物取出倒入玛瑙研钵中进行研磨,得到cd@ba粉末磷光量子产率为 35%。
40.图8是碳纳米点水溶液的紫外-可见吸收光谱和原始样品的rtp激发光谱cd@ba,碳纳米点水溶液在大约247nm和320-390nm处显示出两个主要的吸收带,这分别归因于c=c键的跃迁和氧相关基团的跃迁。原始粒子的rtp激发谱cd@ba显示在365 nm处达到峰值,这与氧相关基团的吸收谱带非常一致。基于这些结果,推测杂原子的掺杂以及ba基质的固定化是观察到的强rtp的主要原因。
41.图9是cd@ba在阴天阳光和365nm光源激发前后的数码照片。在停止紫外或者阴天阳光照射后表现出黄绿色的磷光,在这种环境条件下磷光可持续约9s。
42.实施例5:碳纳米点/硼酸复合磷光材料的制备取碳纳米点溶液1ml(3.4mg/ml)、硼酸ba 3g和29ml去离子水放入烧杯中充分混合,再将烧杯置于200℃的恒温鼓风箱中加热5h,取出后使其快速冷却成块,将烧杯中的块状物取出倒入玛瑙研钵中进行研磨,得到cd@ba粉末磷光量子产率为38%。
43.图10是cd@ba研磨前后的x射线衍射(xrd)光谱,从图中可以看出,在xrd结果中,对于原始cd@ba,观察到较宽的弱峰,这与非晶态一致。研磨后,研磨cd@ba显示出尖锐的衍射峰,表明研磨诱导的从非晶态到晶态的相变。
44.图11是cd@ba研磨前后的差示扫描量热(dsc)曲线,在图中原始的cd@ba在152℃
(90.47j/g)下显示出微弱的吸热峰。而对于研磨后的cd@ba粉末,吸热峰变得尖锐并增强(296.8j/g),表明温度升高的条件下,cd@ba的粉末比原始的cd@ba块状体更稳定,这进一步证实了从高能非晶到低能晶体的转变。
45.实施例6:碳纳米点/硼酸复合磷光材料的制备取碳纳米点溶液1ml(3.4mg/ml)、硼酸ba 3g和29ml去离子水放入烧杯中充分混合,再将烧杯置于150℃的恒温鼓风箱中加热5h,取出后使其快速冷却成块,将烧杯中的块状物取出倒入玛瑙研钵中进行研磨,得到cd@ba粉末磷光量子产率为41%。
46.实施例7:碳纳米点/硼酸复合磷光材料的制备取碳纳米点溶液1ml(3.4mg/ml)、硼酸ba 3g和29ml去离子水放入烧杯中充分混合,再将烧杯置于350℃的恒温鼓风箱中加热3h,取出后使其快速冷却成块,将烧杯中的块状物取出倒入玛瑙研钵中进行研磨,得到cd@ba粉末磷光量子产率为36%。
47.对比例1:取碳纳米点溶液1ml(3.4mg/ml)、硼酸ba 3g和29ml去离子水放入烧杯中充分混合,再将烧杯置于低温环境中蒸发结晶,取出烧杯中的块状物倒入玛瑙研钵中进行研磨,得到蒸发结晶样品(e-cd@ba)。得到的e-cd@ba粉末磷光量子产率为8%。
48.图12是e-cd@ba和cd@ba不同负载浓度的碳纳米点溶液制备出的样品的数码照片。在图中可以清楚地看到,cd@ba样品在日光下是透明的,没有明显的聚集和分层,意味着碳纳米点均匀地分配到硼酸晶体中;而e-cd@ba即使在低负载下也会严重聚集在硼酸晶体的表面。
49.图13是不同负载浓度的碳纳米点溶液下e-cd@ba和cd@ba的磷光强度,随着碳纳米点负载浓度的增加,cd@ba的磷光强度逐渐增加,在浓度为4.5 mg/g时达到最大值;而对于e-cd@ba当浓度为0.3 mg/g时,磷光发射最强。e-cd@ba中碳纳米点更容易在硼酸晶体上或之间聚集,由于晶体中的负载能力较低,导致强度较弱。而对于cd@ba,cds可以最大程度地均匀嵌入硼酸晶体中,因此cd@ba即使在高负载条件下,仍能保持高光学质量。
50.所述实施例为本发明的优选的实施方式,但本发明并不限于上述实施方式,在不背离本发明的实质内容的情况下,本领域技术人员能够做出的任何显而易见的改进、替换或变型均属于本发明的保护范围。